
胞外热休克蛋白70在急性胰腺炎肺损伤中的调节作用
2022-04-07 09:35李影戴胜杰孙学成孔鸿儒陈咨苗孙洪伟
温州医科大学学报 2022年3期
李影,戴胜杰,孙学成,孔鸿儒,陈咨苗,孙洪伟
温州医科大学附属第一医院,浙江 温州 325015,1.手术室;2.肝胆胰外科;3.内分泌科
急性胰腺炎(acute pancreatitis, AP)是临床上常见的急腹症,急性肺损伤是其最常见的并发症和死因[1]。发病时,胰腺自身消化酶激活对胰腺自身产生消化作用,胰腺局部的炎症损伤导致热休克蛋白(heat shock proteins, HSPs)从受损细胞向胞外释放,而后通过血液循环到达全身各处,激活其他细胞表面模式识别受体,促进炎症反应进程[2], 从而破坏血管内皮细胞,引起多脏器炎症连锁反应及功能衰竭(其中肺脏器官最易受损)[3-4]。因此,本研究探讨在AP炎症反应进程中,HSP70是否通过攻击肺血管内皮细胞,造成内皮细胞损伤,从而进一步加重炎症反应。期望寻找有效的炎症反应靶点来遏制AP炎症连锁反应。
1 材料和方法
1.1 主要试剂与抗体 小鼠HSP70 ELISA试剂盒(江苏雨桐生物科技有限公司),TNF-α、IL-6 ELISA试剂盒(美国Proteintech公司),雨蛙素、LPS(北京索莱宝科技有限公司),HSP70抗体(美国Proteintech公司)。
1.2 AP小鼠造模方法及分组 本研究采用雌性NIH小鼠(体质量约25 g),AP造模前禁食18 h,随后连续腹腔注射雨蛙素(50 μg/kg)6次,每次间隔1 h,末次腹腔注射雨蛙素后随即注射脂多糖(10 mg/kg); 对照组小鼠腹腔注射等体积0.9%氯化钠溶液。成功构建对照组小鼠和AP小鼠模型组后,相应对照组(n=6)、AP组(n=6)、HSP70+AP组(n=6)分别通过尾静脉注射3次0.9%氯化钠溶液、0.9%氯化钠溶液和HSP70抗体,从而最终构建本研究采用的3个实验组。
1.3 胰腺组织含水量测定方法 处死小鼠后仔细解剖小鼠胰腺,剔除非胰腺组织,随后分别测量解剖后胰腺组织质量和经60 ℃烘箱烘烤24 h后胰腺组织质量,计算公式:胰腺组织含水量=(胰腺组织湿质量-胰腺组织干质量)/胰腺组织湿质量× 100%。
1.4 HE染色 将不同实验组小鼠解剖后的胰腺组织和肺脏组织置于10%中性甲醛中固定,随后通过石蜡包埋,后将石蜡切片、烤片,经过切片脱蜡至水、苏木素染细胞核、伊红染细胞质、脱水封片等步骤,最后将成型HE染色切片置于显微镜下观察,采集实验图像。
1.5 ELISA实验 收集处死后的不同实验组小鼠血液,留置于红盖管中,室温放置1 h,待血液凝固后置于离心机内,2 000×g离心10 min,随后收集相应上清液,最后按照不同ELISA试剂盒说明书,按步分别检测HSP70、TNF-α和IL-6(其ELISA试剂盒货号分别为MM-0458M1、KE10002和KE10007)。
1.6 转录组测序实验 将不同实验组小鼠肺脏组织进行转录组测序(RNA-seq),检测数据进行基础比对,其基础比对率均>95%,保证样本测序数据库的有效性。差异表达基因(differentially expressed genes, DEGs)分析:通过R软件包EdgeR,并设定显著DEGs筛选阈值为FDR<0.05且fold change>2,筛选不同组之间的显著DEGs;功能富集分析:通过GOKEGG对显著DEGs进行富集分析,纵坐标为富集到的相关通路的具体描述,横坐标为富集比例(公式为富集到基因数除以基因组上该GO类别总注释基因数);基因互相作用网络图:通过富集分析筛选最相关的作用通路,将其中的富集DEGs取交集,通过String数据库分析其间的作用,绘制DEGs互相作用图。
1.7 qRT-PCR实验 为了验证RNA-seq的准确性,我们挑选与炎症密切相关的6个差异DEGs(Fos、FosB、CD274、calcitonin/Ramp2、CD36、NES)进行荧光定量PCR实验,提取不同实验组组织总RNA,并将不同组别RNA进行反转录,加入SYBR Green Master进行RT-PCR,最后使用PCR系统进行qRT-PCR定量分析,计算相应2-ΔΔCt值,并进行分析。GAPDH作为内参。不同基因PCR引物序列见表1。

表1 PCR引物序列(5’-3’)
1.8 统计学处理方法 采用SPSS20.0软件进行相关数据分析,每组数据分别独立重复3次。计量资料以±s表示,多组间比较采用单因素方差分析。P<0.05为差异有统计学意义。
2 结果
2.1 胞外HSP70在AP模型小鼠中起着促炎作用 通过解剖腹腔注射雨蛙素和脂多糖构建的AP模型小鼠和相应试验组小鼠,发现AP组的胰腺表现为最高的胰腺组织含水量,这与AP急性炎症水肿相符合;其次,HSP70+AP组小鼠的胰腺含水量低于AP组,但高于对照组(P<0.05),见图1A,结果初步表明HSP70抗体具有遏制AP进程作用,但不能完全逆转至治愈AP,故可初步推断,胞外HSP70在AP进程中起着促炎作用。HE染色结果显示,AP组中的胰腺和肺脏组织较对照组明显水肿,而HSP70+AP组的胰腺和肺脏组织水肿程度低于AP组,见图1B。
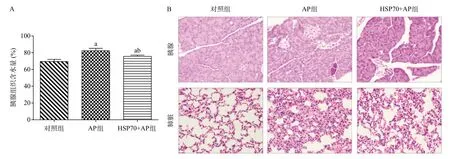
图1 胞外HSP70在AP小鼠模型中的促炎作用
2.2 胞外HSP70表达水平与促炎细胞因子表达水平相关 AP组小鼠的胞外HSP70水平显著升高,而HSP70+AP组小鼠的胞外HSP70水平较AP组降低,但仍高于对照组小鼠(P<0.05);同时,AP组小鼠的TNF-α和IL-6水平亦明显升高,其HSP70+AP组水平较AP组降低,但仍高于对照组小鼠(P<0.05),见图2。表明胞外HSP70在AP进程中具有促进炎症反应的作用,而抑制胞外HSP70水平具有一定抑炎效果。

图2 胞外HSP70及相关细胞促炎因子在各组小鼠中的表达
2.3 胞外HSP70与AP相关肺损伤的细胞黏附能力改变相关 通过DEGs分析可发现,对照组的肺组织在转录组水平上与AP组之间存在最多的DEGs,而AP组与HSP70+AP组之间的DEGs差距最小(见图3A),这初步说明,在AP的炎症进程中,肺组织联动发生了相应的免疫反应。通过对相关DEGs进行功能富集分析,结果显示,DEGs的功能主要集中在细胞黏附功能方面(见图3B)。最后,我们通过筛选显著DEGs高参与的ECM-receptor interaction、Cytokinecytokine receptor interaction、Focal adhesion、 PI3K-Akt信号通路中的差异基因交集,绘制了DEGs互相作用图,为AP炎症进程中胞外HSP70造成的相关基因互作提供了关系图(见图3C)。
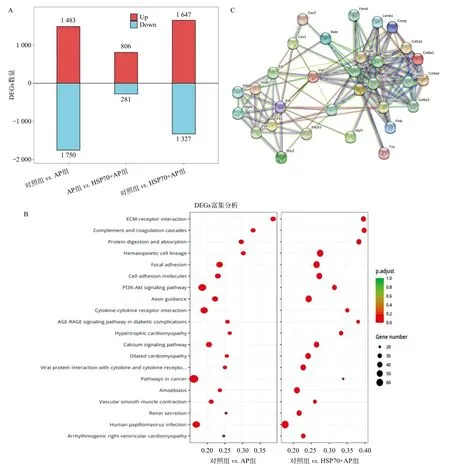
图3 各组小鼠肺组织转录组分析
2.4 荧光定量PCR结果和RNA-seq结果表达趋势一致 为了验证RNA-seq的准确性,我们挑选与炎症密切相关的6个DEGs(Fos,FosB,CD274,calcitonin/ Ramp2,CD36,NES)进行荧光定量PCR实验。结果显示,在AP组中,CD36、NES、Calcitonin(Ramp2)的表达量显著上升,而Fos、FosB和CD274表达量下降,见图4,表明荧光定量PCR结果和RNA-seq结果表达趋势一致,转录组测序结果稳定可靠。

图4 各组中显著DEGs的mRNA相对表达水平
3 讨论
AP作为一种急性起病的胰腺相关的全身性炎症反应性疾病,其病情凶险、病程长、病死率较高。在AP发展过程中,其炎症级联反应往往不仅局限于胰腺器官本身,常伴随着机体其他远处器官的功能衰竭,如急性肾功能不全、急性呼吸窘迫综合征等,其中又以急性肺损伤最为常见。急性肺损伤引发的肺泡毛细血管弥漫性损伤往往导致肺水肿和肺不张,临床症状主要为呼吸窘迫和顽固性低氧血症,这又给AP的治疗带了阻碍[5]。本研究期望阻断AP炎症反应进程中的急性肺损伤来改善AP的预后。
HSPs在蛋白质的折叠、转运、生物合成等方面扮演着重要角色,而机体应激时,大量HSPs释放入血,参与全身性炎症反应过程。胞外HSP70作为重要的炎症介导因子之一,在释放入血后可能会引起胰外器官的炎症损伤,这将是在AP发展进程中阻断急性肺损伤发生极为重要的一环。本研究结果显示,AP小鼠在经HSP70抗体处理后,人为地降低胞外HSP70表达水平,其胰腺组织和肺脏组织损伤程度较AP组明显降低,同时TNF-α、IL-6等促炎因子浓度呈恶性循环式加重的现象也得到了明显遏制。基于上述结果表现,我们初步推测在AP炎症反应过程中,胞外HSP70可能通过影响某些信号通路而加重AP肺损伤,抑制胞外HSP70可以明显改善AP的肺损伤。
为了探讨胞外HSP70发挥促进炎症反应作用的分子机制,将不同实验组别肺组织进行转录组测序分析,结果表明:AP组与治疗组之间的差异较小,说明有些免疫反应是共同发生的,只有部分的免疫反应是某一组特异的。DEGs的功能富集分析结果表明:DEGs显著富集的通路有ECM-receptor interaction, Cytokine-cytokine receptor interaction,Focal adhesion,PI3K/Akt等通路,这些通路都与细胞的黏附、炎症反应相关。进一步的qRT-PCR结果验证转录组测序结果,表达差异最显著基因的改变趋势与转录组测序结果一致,其中CD36水平亦在经HSP70抗体处理后的AP组中较AP组下降,CD36可作为TLR4的辅助受体介导脂多糖相关的炎症反应,也可参与mTORC1信号通路中调控炎症反应[6-8]。相应地,Fos、FosB等分子表达水平亦与PI3K/Akt/mTOR信号通路密切相关。因此,我们推测,胞外HSP70通过影响ECM-receptor interaction,Cytokine-cytokine receptor interaction,Focal adhesion,PI3K/Akt等信号通路而加重炎症损伤导致肺脏组织内皮细胞受损,从而达到诱发AP过程中急性肺损伤的作用。
在AP发展中,HSP70被大量释放入血,通过影响ECM-receptor interaction,Cytokine-cytokine receptor interaction,Focal adhesion,PI3K/Akt等信号通路,导致大量炎性细胞因子释放,损害内皮细胞屏障,导致全身多脏器损伤,同时炎症又反过来刺激释放HSP70,最终导致AP患者的病情极速恶化,从而导致不良预后。假如能有效地阻断胞外HSP70释放,进而保护内皮细胞屏障不被破坏,将有效抑制AP发展过程中多脏器功能衰竭的恶化进程,故继续深入研究胞外HSP70在AP发生发展进程中的具体机制,对AP的治疗将产生巨大的临床应用价值。但本研究仅研究了胞外HSP70促进AP并发急性肺损伤的表象,初步探讨了它发挥上述作用过程中可能影响的信号通路,未对其详细的分子机制进行验证,在接下来的研究中将选择前述的几条信号通路进行逐一验证,期待能揭示胞外HSP70诱发AP肺损伤的具体机制。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
中国CT和MRI杂志(2022年10期)2022-10-18
中国典型病例大全(2022年7期)2022-04-22
健康之家(2021年6期)2021-09-08
科学导报(2021年29期)2021-06-03
人人健康(2020年4期)2020-05-25
中国现代医生(2019年18期)2019-09-02
科海故事博览·下旬刊(2019年6期)2019-04-16
恋爱婚姻家庭·养生版(2018年6期)2018-08-01
农家科技(2018年4期)2018-06-23
