
有氧间歇运动对2型糖尿病大鼠心肌线粒体自噬的影响*
2022-03-28 05:59侯改霞习雪峰刘倩倩付素焕唐书曼卞寒雪
中国病理生理杂志 2022年3期
侯改霞, 习雪峰, 刘倩倩, 付素焕, 唐书曼, 卞寒雪
有氧间歇运动对2型糖尿病大鼠心肌线粒体自噬的影响*
侯改霞, 习雪峰△, 刘倩倩, 付素焕, 唐书曼, 卞寒雪
(河南大学体育学院,河南 开封 475001)
探讨线粒体自噬在运动(Exe)干预2型糖尿病心肌病中的作用及可能机制。自发性2型糖尿病Goto-Kakizaki (GK)大鼠随机分为2组:糖尿病组(GK组)和GK+Exe组,每组8只;另增设8只同周龄雄性Wistar大鼠作为对照组(Wistar组)。GK+Exe组大鼠经有氧间歇运动干预8周,每天60 min,每周5 d。在最后一次运动结束48 h后,过夜禁食12 h后,将大鼠腹腔麻醉、剖腹,腹主动脉采血,检测空腹血糖(FBG)、空腹胰岛素(FINS)及血清乳酸脱氢酶(LDH)和肌酸激酶(CK)水平。制作心肌透射电子显微镜切片,观察心肌细胞及线粒体的超微结构。Western blot法测定心肌组织中p-AMPK、p-ULK1、TFEB、PINK1、parkin、LC3-II、LC3-I和p62蛋白表达,并计算LC3-II/LC3-I比值。与Wistar组相比,GK组大鼠FBG及血清LDH和CK水平显著升高(<0.01),FINS水平显著降低(<0.01);心肌线粒体结构欠缺,有空泡现象,偶见自噬体;心肌p-ULK1表达显著下降(<0.01),p62表达显著升高(<0.01)。与GK组相比,GK+Exe组大鼠FBG和血清CK水平显著降低(<0.05),FINS水平显著升高(<0.05);心肌线粒体结构基本完整,空泡现象减轻,自噬体形成增多;心肌p-AMPK、p-ULK1、TFEB和parkin表达及LC3-II/LC3-I显著升高(<0.05或<0.01),p62表达显著下降(<0.05)。有氧间歇运动对2型糖尿病心肌病具有一定的缓解作用,其机制可能与激活心肌AMPK/ULK1信号通路,提高心肌线粒体自噬有关。
糖尿病心肌病;有氧间歇运动;线粒体自噬;AMPK/ULK1信号通路
2型糖尿病是一种以胰岛素分泌和利用缺陷为特征的慢性代谢性疾病。糖尿病患者发生心力衰竭的风险(39%)是非糖尿病患者(23%)的近两倍,心力衰竭是糖尿病患者死亡的主要原因[1]。线粒体对于心肌收缩、电稳定性、细胞存活和凋亡非常重要。在高血糖环境下,心肌细胞内受损/缺陷线粒体积累增多,心肌细胞代谢和功能异常[2]。线粒体自噬可选择性的清除衰老或损伤的线粒体,维持心肌细胞线粒体的数量与形态平衡,达到抑制氧化应激、减少细胞损伤的作用。因此,线粒体自噬与糖尿病心肌病的发生、发展密切相关。
AMPK(AMP-activated protein kinase)及其下游调节因子ULK1(Unc-51 like autophagy activating kinase 1)已被证明在线粒体自噬中发挥关键作用。运动是防治2型糖尿病及其并发症的常用辅助疗法之一。运动(exercise, Exe)可通过激活AMPK,提高运动后骨骼肌线粒体自噬水平;若抑制AMPK活性或敲除基因,运动则无法诱导骨骼肌细胞线粒体自噬的变化[3]。运动可有效诱导自噬的发生,一次急性运动可使心肌细胞内自噬相关蛋白的表达发生变化,运动对心肌的保护作用部分是通过运动增加自噬实现的[4],但具体机制还有待于进一步研究。已有研究表明,运动对糖尿病心肌病确有一定的缓解作用[5-6]。但是,关于线粒体自噬在运动缓解糖尿病心肌病中作用和机制的研究还较少。因此,本研究将以AMPK/ULK1信号通路作为出发点,探讨线粒体自噬在运动干预2型糖尿病心肌病中的作用及可能机制。
材料和方法
1 材料
18只自发性2型糖尿病Goto-Kakizaki (GK)大鼠(300~350 g)和8只Wistar大鼠(400~450 g)均购自斯贝福(北京)生物技术有限公司[16周龄,雄性,SPF级,许可证号为SCXK(京)2019-0010]。
乳酸脱氢酶(lactate dehydrogenase, LDH)和肌酸激酶(creatine kinase, CK)检测试剂盒(南京建成生物工程研究所);胰岛素ELISA试剂盒(联科生物);兔抗大鼠p-AMPK多克隆抗体(CST);兔抗大鼠p-ULK1多克隆抗体和小鼠抗大鼠PINK多克隆抗体(Abcam);兔抗大鼠parkin多克隆抗体和小鼠抗大鼠GAPDH多克隆抗体(Servicebio);兔抗大鼠微管相关蛋白1轻链3(microtubule-associated protein 1 light chain 3, LC3)多克隆抗体(武汉三鹰);小鼠抗大鼠p62单克隆抗体(Santa);兔抗大鼠转录因子EB(transcription factor EB, TFEB)多克隆抗体(BIOSS);其他试剂均为国产分析纯。
透射电子显微镜(HITACHI);台式高速冷冻型微量离心机(Dragon Lab);酶标检测仪(Rayto);电泳仪(北京六一仪器厂);图像采集分析系统(Nikon);血糖仪(日本京都);动物跑台(成都泰盟)。
2 方法
2.1实验分组GK大鼠经过1周的适应性喂养后,检测随机血糖,并进行口服糖耐量实验。GK大鼠随机血糖≥11.1 mmol/L且口服糖耐量降低为2型糖尿病GK大鼠成模标准[7-8],共筛选出16只GK大鼠用于后续实验。将筛选出的GK大鼠随机分为2组:糖尿病组(GK组)和GK+Exe组,每组8只。另设8只同周龄雄性Wistar大鼠作为对照组(Wistar组)。所有大鼠分笼饲养,自由饮水、摄食,室温为(22±2) ℃,相对湿度为40%~60%,光照/黑暗周期为12 h(8:00~20:00)。
2.2运动干预[9]GK+Exe组大鼠采用有氧间歇跑台运动(aerobic interval training, AIT)。正式运动前先进行1周的适应性运动。第2周开始进行正式AIT,每天60 min,每周5 d,共8周。正式运动时,首先进行10 min的热身运动,然后在22 m/min、7 min和15 m/min、3 min之间交替进行共60 min的运动。
2.3实验取材为了避免急性运动对实验结果的影响,在最后一次跑台运动结束48 h后,过夜禁食12 h,采用血糖仪检测大鼠空腹血糖(fast blood glucose, FBG),将大鼠腹腔麻醉、剖腹,腹主动脉采血,全血3 000 r/min离心20 min,取血清置于-80 ℃冰箱保存,用于检测空腹胰岛素(fasting insulin, FINS)、LDH和CK。取部分左心室置于2.5%戊二醛固定液,以备心肌透射电镜切片的制作及观察;另取部分左心室置于-80℃冰箱保存,用于测定心肌线粒体自噬相关蛋白的表达情况。
2.4血液指标的检测FINS、LDH和CK的检测完全按照试剂盒说明书进行。
2.5心肌透射电镜制片及观察(1)固定:取大鼠左心室1 mm×1 mm×1 mm大小的组织置于电镜固定液进行固定。(2)后固定:1%锇酸室温固定2 h。(3)脱水:依次放入30%、50%、70%、80%、95%、100%和100%乙醇脱水,每次20 min,100%丙酮两次,每次15 min。(4)渗透包埋:丙酮∶包埋剂=1∶1, 2~4 h, 丙酮∶包埋剂=1∶2,渗透过夜,纯包埋剂,5~8 h,均在37 ℃条件下进行。(5)聚合:置于60 ℃烤箱聚合48 h。(6)超薄切片:60~80 nm超薄切片,150目铜网捞片。(7)染色:铜网置于醋酸铀饱和乙醇溶液避光染色8 min,枸橼酸铅溶液避二氧化碳染色8 min。(8)透射电镜下观察,采集图像分析。
2.6Western blot检测取100 μg心肌组织加入1 mL裂解液充分裂解,离心,收集上清。BCA法测定蛋白浓度,SDS-PAGE分离,PVDF膜转膜,5%脱脂牛奶封闭,4 ℃孵育Ⅰ抗(1∶1 000)过夜,孵育Ⅱ抗(1∶5 000)30 min,化学发光法显影、定影,扫描胶片存档。以GAPDH为内参照。采用ImageJ软件对实验结果进行半定量分析。
3 统计学处理
采用SPSS 20.0软件包进行统计分析。数据用均数±标准差(mean±SD)表示。组间比较采用单因素方差分析。以<0.05为差异有统计学意义。
结果
1 大鼠的一般体征
Wistar组大鼠体形正常,皮毛呈白色且有光泽,饮食、大小便正常,精神状态良好,反应敏捷;GK组大鼠体型较瘦,毛色发黄,缺少光泽,反应迟钝,出现多饮多尿症状;GK+Exe组大鼠体形、皮毛色泽、精神状态、反应等情况均有一定程度的改善。
2 大鼠血液相关指标的比较
如表1所示,与Wistar组相比,GK组大鼠FBG、LDH和CK水平显著升高(<0.01),FINS水平显著降低(<0.01);与GK组相比,GK+Exe组大鼠FBG和CK水平显著降低(<0.05),FINS水平显著升高(<0.05),但GK+Exe组大鼠FBG和LDH水平仍显著高于Wistar组(<0.01),FINS水平显著低于Wistar组(<0.01)。

表1 不同组别大鼠FBG、FINS、LDH和CK水平的比较
3 大鼠心肌超微结构的比较
如图1所示,Wistar组大鼠心肌线粒体结构完整,无空泡、无肿胀变形,基质致密,线粒体嵴排列整齐、无断裂;GK组大鼠部分线粒体结构欠缺,线粒体嵴部分断裂,有空泡现象,基质密度降低,偶见自噬体;GK+Exe组大鼠线粒体结构基本完整,空泡现象减轻,线粒体基质致密,膜基本完整,自噬体形成增多。
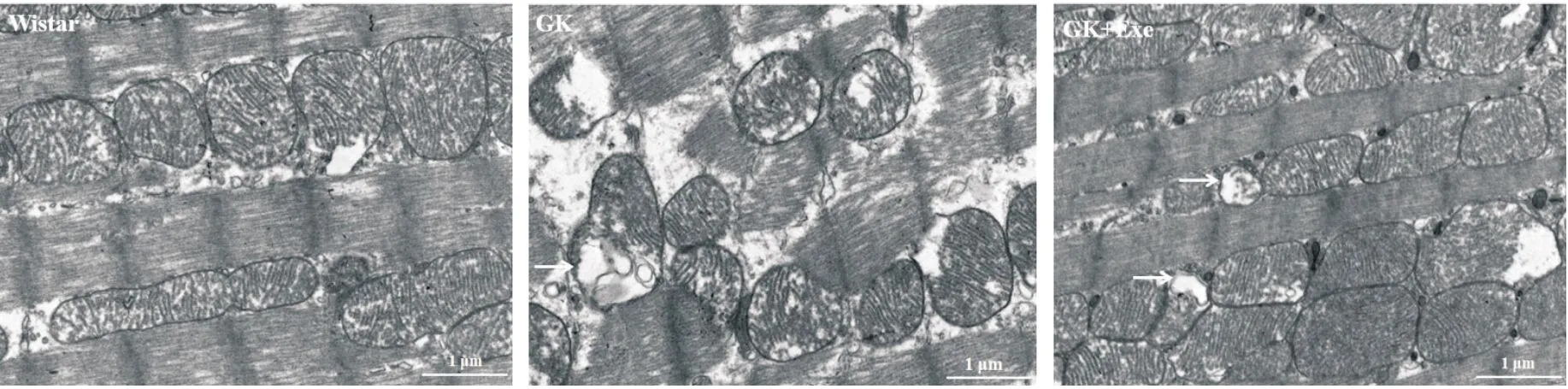
Figure 1.Ultrastructure of myocardial mitochondria in the rats of different groups (×6 000, scale bar=1 μm).
4 大鼠心肌p-AMPK和p-ULK1水平的变化
与Wistar组相比,GK组大鼠心肌p-ULK1水平显著下降(<0.01);与GK组相比,GK+Exe组p-AMPK和p-ULK1水平均显著升高(<0.01),见图2。
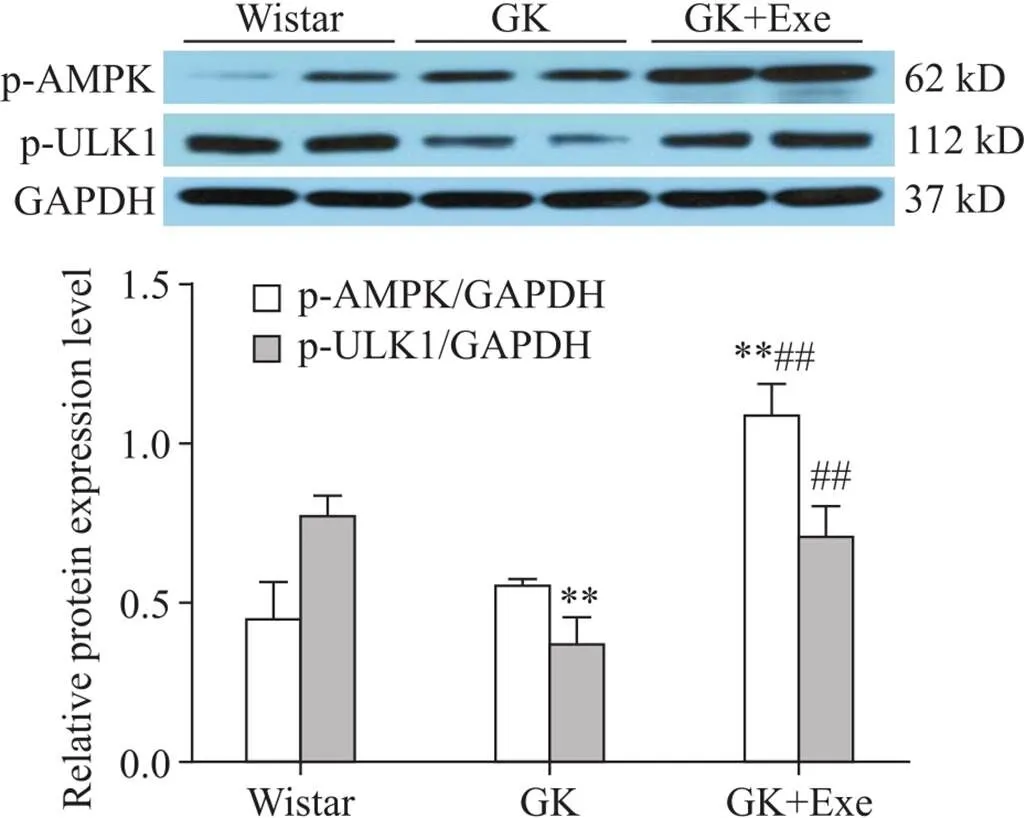
Figire 2.Comparison of expression of p-AMPK and p-ULK1 in myocardium of rats in different groups. Mean±SD. n=8. **P<0.01 vs Wistar group; ##P<0.01 vs GK group.
5 大鼠心肌TFEB表达的变化
与Wistar组相比,GK组大鼠心肌TFEB表达无显著差异(>0.05);与Wistar组和GK组相比,GK+Exe组TFEB表达显著升高(<0.05或<0.01),见图3。
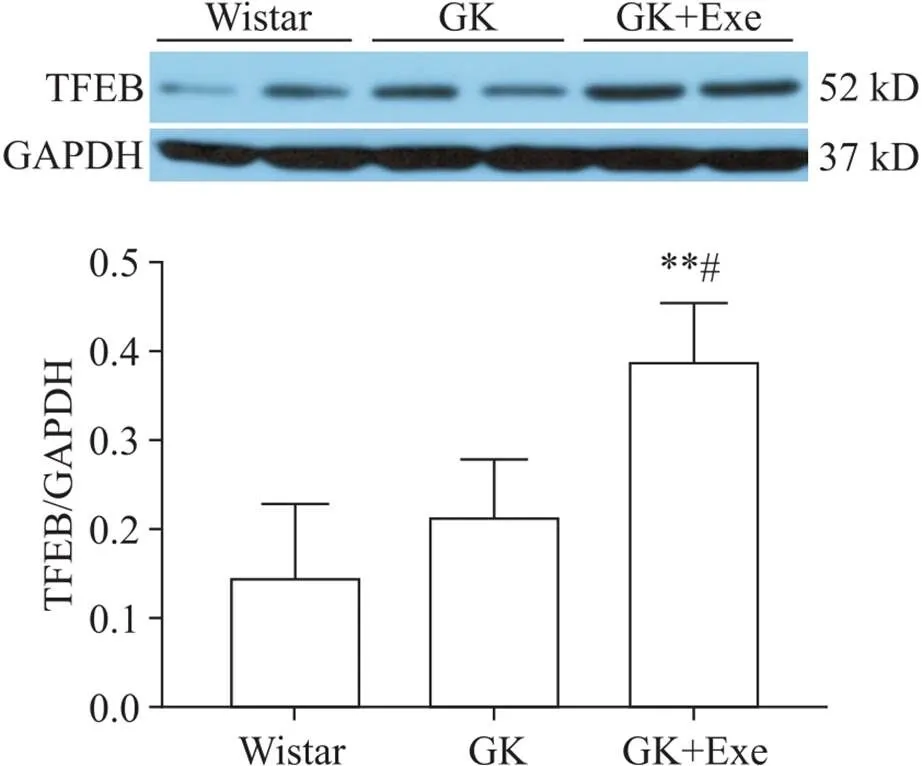
Figure 3.Comparison of expression of TFEB in myocardium of rats in different groups. Mean±SD. n=8. **P<0.01 vs Wistar group; #P<0.05 vs GK group.
6 大鼠心肌PINK1和parkin表达的变化
与Wistar组相比,GK组大鼠心肌PINK1和parkin表达无显著差异(>0.05);与Wistar组相比,GK+Exe组大鼠心肌PINK1表达显著升高(<0.05);与Wistar组和GK组相比,GK+Exe组大鼠心肌parkin表达显著升高(<0.01),见图4。
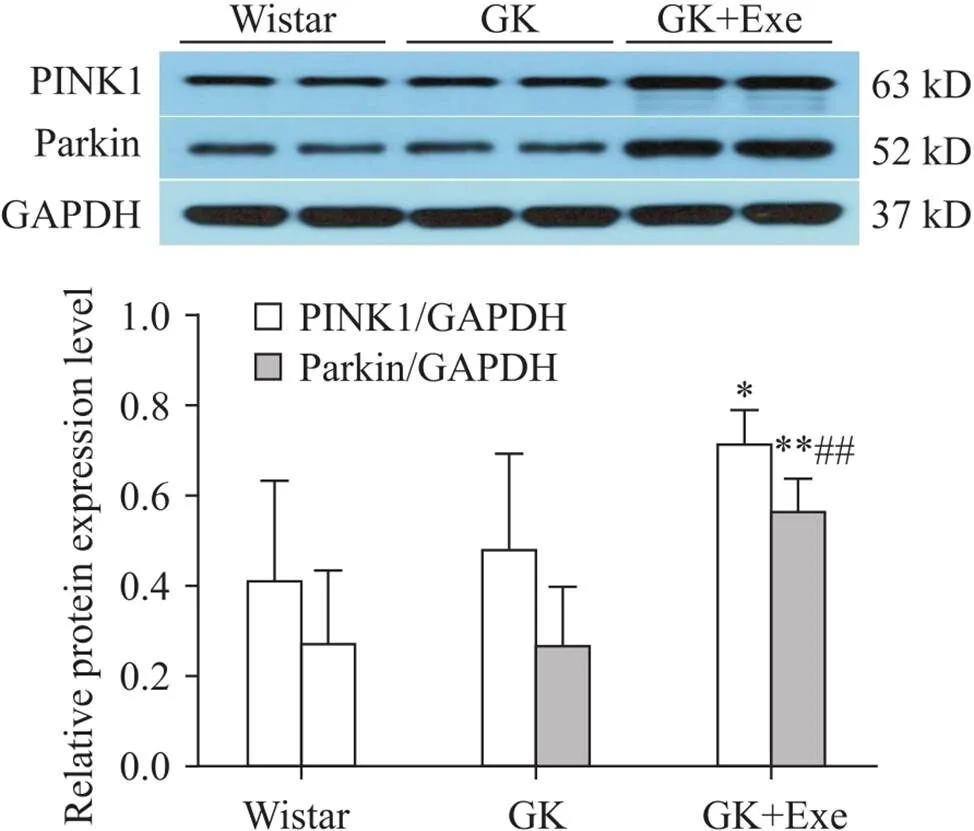
Figure 4.Comparison of expression of PINK1 and parkin in myocardium of rats in different groups. Mean±SD. n=8. *P<0.05, **P<0. 01 vs Wistar group; ##P<0.01 vs GK group.
7 大鼠心肌LC3-II/LC3-I和p62表达的变化
与Wistar组相比,GK组和GK+Exe组大鼠心肌p62表达显著升高(<0.05或<0.01);与GK组相比,GK+Exe组大鼠心肌LC3-II/LC3-I显著升高(<0.01),p62表达显著下降(<0.05),见图5。
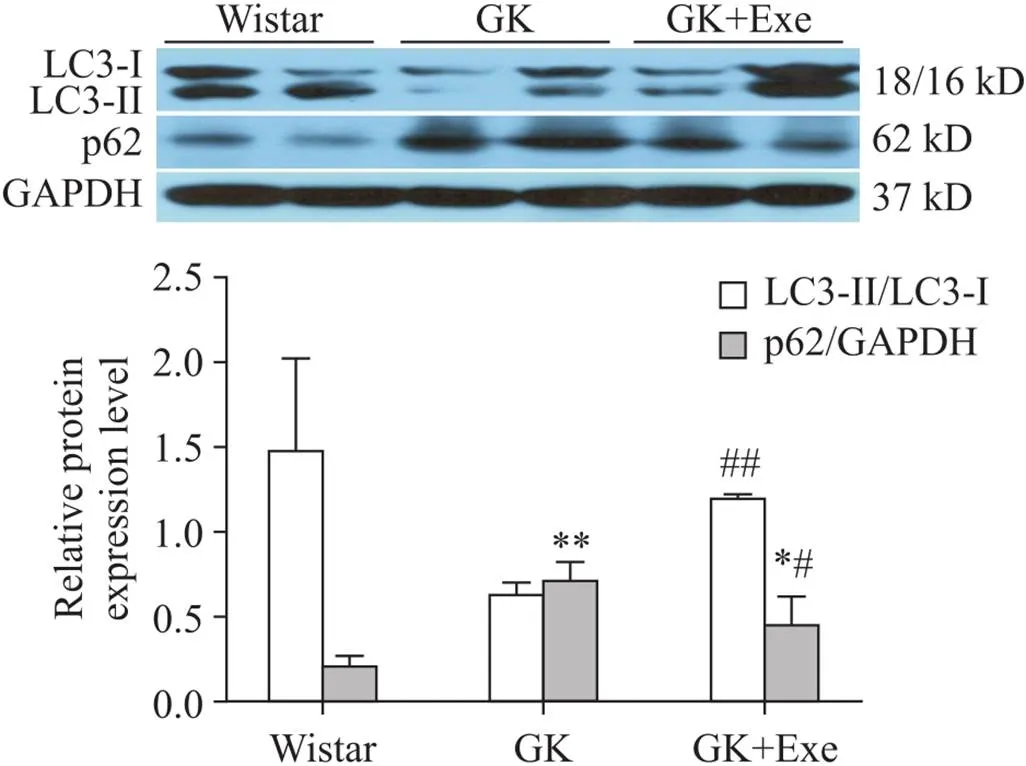
Figure 5.Comparison of expression of p62 and LC3-II/LC3-I in myocardium of rats in different groups. Mean±SD. n=8. *P<0.05,**P<0.01 vs Wistar group; #P<0.05, ##P<0.01 vs GK group.
讨论
线粒体是心肌细胞的“能量工厂”,其体积占据细胞质体积的35%~40%,为心脏跳动提供95%的ATP。在心肌细胞中,线粒体对于心肌收缩、电稳定性和细胞存活或凋亡非常重要,在生理条件和病理应激损伤中调节细胞的多种生理过程[10]。在糖尿病机体中,健康的线粒体对于心肌细胞存活至关重要。然而,在持续的高血糖刺激下,心肌细胞的线粒体断裂、活性氧(reactive oxygen species, ROS)增多,线粒体膜电位和电子传递链活性降低,心肌细胞能量缺乏,心肌细胞凋亡,最终导致心功能不全。线粒体功能障碍可能是糖尿病心肌病的中心介质[11]。在本研究中,2型糖尿病GK大鼠FBG及血清CK和LDH水平显著升高,FINS水平显著降低,电镜观察结果显示心肌线粒体损伤严重;而规律的有氧间歇运动降低了GK大鼠FBG和血清CK水平,升高了FINS水平,心肌细胞线粒体自噬体增多,线粒体损伤减轻。这提示2型糖尿病GK大鼠心肌细胞遭到了一定程度的损害,运动对其缓解作用除了与降低血糖、提高胰岛素水平相关外,可能与增强心肌细胞线粒体自噬,降低线粒体的损伤相关。
线粒体自噬可选择性降解受损或衰老的线粒体,并使其循环利用[12]。以往研究表明,在代谢综合征心脏中,线粒体自噬要么下调[13],要么上调[14]。线粒体自噬在糖尿病心肌病中的潜在机制尚未完全明确。Tong等[15]发现,在糖尿病心肌病早期发展过程中,线粒体自噬被激活,但不足以保护心脏。增强线粒体自噬功能可减轻线粒体功能障碍,减少脂质积聚,并防止心脏舒张功能障碍。通过药物(如非诺贝特、二甲双胍和白藜芦醇等)干预激活自噬可挽救糖尿病引起的心脏功能障碍,而抑制自噬会加剧糖尿病心肌病。
AMPK和ULK1已被证明在线粒体自噬中发挥关键作用[16]。线粒体受损时,线粒体中AMPK表达量约增加3倍,表明AMPK被招募到线粒体以应对线粒体损伤。AMPK可与ULK1富含丝氨酸/脯氨酸的区域相互作用,并直接磷酸化ULK1的多个位点,导致ULK1构象变化,从而促进ULK1与其复合物其他组分的相互作用,增加ULK1活性和稳定性。在基因敲除的小鼠胚胎成纤维细胞,ULK1不能被激活,自噬被抑制,受损线粒体积聚,证明AMPK诱导的ULK1磷酸化在自噬中十分重要[17]。本研究发现,2型糖尿病GK大鼠心肌p-ULK1水平下降,而运动可提高GK大鼠心肌p-AMPK和p-ULK1水平,提示运动可能通过提高GK大鼠心肌p-AMPK和p-ULK1的水平,开启增强心肌线粒体自噬的通路。前人关于运动与骨骼肌线粒体自噬的研究也证实了这一点。急性运动可刺激AMPK和ULK1磷酸化[18],AMPK是运动诱导ULK1特异性磷酸化参与下游线粒体自噬途径所必需的。同样,骨骼肌基因缺失也会抑制线粒体自噬[3],而且ULK1在将溶酶体靶向受损/功能失调的线粒体方面具有重要作用。
TFEB是自噬的主要转录调节因子,可协调约60种溶酶体水解酶以及溶酶体和自噬体膜相关辅助蛋白的表达,同时也调节自噬囊泡的形成、定位和通量[19],激活整个自噬溶酶体途径。本研究发现,运动提高了GK大鼠心肌TFEB表达,这可能与运动提高了GK大鼠心肌p-AMPK水平有关。激活AMPK可通过多种途径调节TFEB表达[20]。TFEB可上调内皮细胞的自噬相关基因的表达,通过促进血管生成,改善心肌梗死后心脏功能的恢复[21],TFEB的下调会导致心肌自噬通量受损、氧化应激增加[22]。
在线粒体自噬过程中,PINK1/parkin信号通路的主要功能为监控线粒体质量,识别受损线粒体,标记受损线粒体,募集自噬受体,开启自噬过程。本研究结果表明,间歇性有氧运动可增强2型糖尿病大鼠心肌PINK1和parkin表达,与提高p-AMPK、p-ULK1和TFEB水平的作用一致,可能在增强糖尿病心肌线粒体自噬方面起到相辅相成的作用。现在研究认为,AMPK和(或)ULK1的一些下游靶点可能参与PINK1/parkin信号通路。而parkin中ULK1磷酸化位点突变、遗传性或基因缺失或药理学ULK1抑制,都会延迟parkin的激活,导致parkin和线粒体自噬功能的缺陷[16]。同样,TFEB与PINK1/parkin诱导的线粒体自噬也有关系。PINK1/parkin与线粒体结合依赖于TFEB[23]。反过来,PINK1/parkin也可促进TFEB向细胞核的移位,激活溶酶体基因的表达,增强线粒体自噬[24]。
磷脂酰乙醇胺(phosphatidylethanolamine, PE)是自噬小体的一种整合蛋白,Atg8家族蛋白与PE的结合是吞噬囊泡形成的一个关键因素。LC3是Atg8蛋白中的亚家族,通常以LC3-I的形式存在于细胞质中。当线粒体自噬发生时,LC3-I通过类泛素样修饰的方式与PE共价结合,形成LC3-II。此后,LC3-II促进自噬泡双层膜的起始、延伸及闭合,提供自噬受体的结合位点,并与溶酶体膜融合。LC3-II与自噬小体数量紧密相关,是反映线粒体自噬活性的关键指标。p62通过其LIR(LC3-interaction region)结构域与LC3形成复合物,被包裹进自噬小体,可作为自噬特异性底物在自噬溶酶体内降解[25]。自噬发生时,p62被不断降解;当自噬功能受损或减弱时,p62蛋白明显累积。因此,p62蛋白表达水平与自噬活性成反比,是检测线粒体自噬的一个辅助指标。本研究结果表明,间歇性有氧运动可提高2型糖尿病大鼠心肌LC3-II/LC3-I水平和p62蛋白表达,从而促进心肌线粒体自噬。研究发现,活化的AMPK可以调节ULK1磷酸化,进而磷酸化FUN14结构域包含蛋白1(FUN14 domain containing 1, FUNDC1)的Ser17位点,提髙FUNDC1与LC3的结合效率,促进线粒体自噬[26]。此外,磷酸化ULK1还可通过对p62的UBA结构域Ser403位点的磷酸化作用,导致p62泛素化,激活线粒体自噬[27]。
总之,鉴于上述的AMPK/ULK1与TFEB、PINK1/parkin、LC3和p62在线粒体自噬中的密切关系,结合本文的研究结果,提示间歇性有氧运动可能通过增强2型糖尿病大鼠心肌AMPK/ULK1信号通路的作用,提高了心肌线粒体自噬,从而缓解2型糖尿病心肌病。然而,具体机制仍有待进一步研究。
[1] Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitus[J]. Circ Res, 2019, 124(1):121-141.
[2] Gollmer J, Zirlik A, Bugger H. Mitochondrial mechanisms in diabetic cardiomyopathy[J]. Diabetes Metab J, 2020, 44(1):33-53.
[3] Laker RC, Drake JC, Wilson RJ, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy[J]. Nat Commun, 2017, 8(1):548.
[4] Gobbidi S, Laher I. Molecular mechanisms in exercise induced cardioprotection[J]. Cardiol Res Pract, 2011, 2011:972807.
[5] Novoa U, Arauna D, Moran M, et al. High-intensity exercise reduces cardiac fibrosis and hypertrophy but does not restore the nitroso-redox imbalance in diabetic cardiomyopathy[J]. Oxid Med Cell Longev, 2017, 2017:7921363.
[6] Silva FS, Bortolin RH, Araújo DN, et al. Exercise training ameliorates matrix metalloproteinases 2 and 9 messenger RNA expression and mitigates adverse left ventricular remodeling in streptozotocin-induced diabetic rats[J]. Cardiovasc Pathol, 2017, 29:37-44.
[7] Zhang N, Zheng Q, Wang Y, et al. Renoprotective effect of the recombinant anti-IL-6R fusion proteins by inhibiting JAK2/STAT3 signaling pathway in diabetic nephropathy[J]. Front Pharmacol, 2021, 12:681424.
[8] Berdugo M, Delaunay K, Lebon C, et al. Long-term oral treatment with non-hypoglycemic dose of glibenclamide reduces diabetic retinopathy damage in the Goto-Kakizaki rat model[J]. Pharmaceutics, 2021, 13(7):1095.
[9] Yue X, Gong DW, Tian Z. FSTL1 as a potential mediator of exercise-induced cardioprotection in post-myocardial infarction rats[J]. Sci Rep, 2016, 6:32424.
[10] Tahrir FG, Langford D, Amini S, et al. Mitochondrial quality control in cardiac cells: mechanisms and role in cardiac cell injury and disease[J]. J Cell Physiol, 2019, 234(6):8122-8133.
[11] Gollmer J, Zirlik A, Bugger H. Mitochondrial mechanisms in diabetic cardiomyopathy[J]. Diabetes Metab J, 2020, 44(1):33-53.
[12] Bharath LP, Rockhold JD, Conway R. Selective autophagy in hyperglycemia-induced microvascular and macrovascular diseases[J]. Cells, 2021, 10(8):2114.
[13] Kanamori H, Takemura G, Goto K, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes[J]. Autophagy, 2015, 11(7):1146-1160.
[14] Russo SB, Baicu CF, Van Laer A, et al. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes[J]. J Cin Invest, 2012, 122(11):3919-3930.
[15] Tong M, Saito T, Zhai P, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy[J]. Circ Res, 2019, 124(9):1360-1371.
[16] Hung CM, Lombardo PS, Malik N, et al. AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy[J]. Sci Adv, 2021, 7(15):eabg4544.
[17] Li Y, Chen Y. AMPK and autophagy[J]. Adv Exp Med Biol, 2019, 1206:85-108.
[18] Møller AB, Vendelbo MH, Christensen B, et al. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle[J]. J Appl Physiol (1985), 2015, 118(8):971-979.
[19] Astanina E, Bussolino F, Doronzo G. Multifaceted activities of transcription factor EB in cancer onset and progression[J]. Mol Oncol, 2020, 15(2):327-346.
[20] Li X, Yu W, Qian X, et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy[J]. Mol Cell, 2017, 66(5):684-697.
[21] Fan YB, Lu HC, Liang WY, et al. Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis[J]. Circ Res, 2018, 122(7):945-957.
[22] Liu H, Liu S, Qiu X, et al. Donor MSCs release apoptotic bodies to improve myocardial infarction via autophagy regulation in recipient cells[J]. Autophagy, 2020, 16(12):2140-2155.
[23] Liu W, Li CC, Lu X, et al. Overexpression of transcription factor EB regulates mitochondrial autophagy to protect lipopolysaccharide-induced acute lung injury[J]. Chin Med J (Engl), 2019, 132(11):1298-1304.
[24] Zhang Y, Nguyen DT, Olzomer EM, et al. Rescue of Pink1 deficiency by stress-dependent activationof autophagy[J]. Cell Chem Biol, 2017, 24 (4):471-480.
[25] Xu Y, Shen J, Ran J. Emerging views of mitophagy in immunity and autoimmune diseases[J]. Autophagy, 2020,16(1):3-17.
[26] Zhang T,Liu Q , GAO W, et al. The multifaceted regulation of mitophagy by endogenous metabolites[J/OL]. Autophagy, 2021 (2021-09-09) [2021-11-25]. https://www.tandfonline.com/doi/full/10.1080/15548627.2021. 1975914.
[27] Lee DH, Park JS, Lee YS, et al. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity[J]. Autophagy, 2020, 16(11):1949-1973.
Effects of aerobic interval training on autophagy of myocardial mitochondria in type 2 diabetic rats
HOU Gai-xia, XI Xue-feng△, LIU Qian-qian, FU Su-huan, TANG Shu-man, BIAN Han-xue
(,,475001,)
To investigate the effect of exercise (Exe) intervention on type 2 diabetic cardiomyopathy and its possible mechanism.Spontaneously type 2 diabetic Goto-Kakizaki (GK) rats were randomly divided into diabetic group (GK group) and GK+Exe group, while another 8 male Wistar rats served as control group (Wistar group). The rats in GK+Exe group were treated with aerobic interval training, 60 min per day, 5 d per week for 8 weeks. The rats were anesthetized, and the blood was collected 48 h after the last Exe with overnight fasting. Fasting blood glucose (FBG), fasting inslulin (FINS), and serum LDH and CK levels were measured. Ultrastructure of cardiomyocytes and mitochondria was observed by transmission electron microscopy. The protein levels of p-AMPK, p-ULK1, TFEB, PINK1, parkin, LC3-II, LC3-I and p62 in myocardial tissues were measured by Western blot, and the ratio of LC3-II/LC3-I was calculated.Compared with Wistar group, the FBG, and serum LDH and CK levels were increased in GK group (0.01), and FINS in GK group was decreased significantly (<0.01). Incomplete mitochondrial structure, vacuole and occasional autophagy were observed in the myocardial mitochondria of GK rats. The expression of p-ULK1 was decreased, while p62 was increased in GK group (0.01). Compared with GK group, the FBG and serum CK levels were decreased, while FINS was increased significantly in GK+Exe group (0.05). Normal mitochondrial structure, less vacuoles and more autophagosomes were observed in GK+Exe group. Exe also elevated the levels of p-AMPK, p-ULK1, TFEB, parkin and LC3-II/LC3-I, but inhibited p62 expression (0.05).Aerobic interval training attenuates type 2 diabetic cardiomyopathy through AMPK/ULK1 signaling pathway and myocardial mitochondrial autophagy.
Diabetic cardiomyopathy; Aerobic interval training; Mitophagy; AMPK/ULK1 signaling pathway
R587.2; R363.2
A
10.3969/j.issn.1000-4718.2022.03.008
**0.01Wistar group;△<0.05GK group.
1000-4718(2022)03-0442-06
2021-12-01
2022-01-20
[基金项目]河南省重点研发与推广专项(科技攻关)项目(No. 202102310318; No. 212102310262);河南省高等学校重点科研项目(No. 20A890002)
Tel: 0378-22866474; E-mail: 271070602@qq.com
(责任编辑:卢萍,罗森)
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
心肺血管病杂志(2020年5期)2021-01-14
中华养生保健(2020年4期)2020-11-16
保健文汇(2020年7期)2020-08-21
保健与生活(2020年13期)2020-07-24
体育科学(2018年12期)2019-01-04
分析化学(2017年12期)2017-12-25
科学中国人(2016年9期)2016-11-04
