
电催化分解氨制氢研究进展
2022-03-24 09:16:16王中华郑淞生姚育栋陈日懿王兆林
化工学报 2022年3期
王中华,郑淞生,姚育栋,陈日懿,王兆林
(1厦门大学能源学院,福建 厦门 361102; 2中国三峡新能源(集团)股份有限公司,北京 101100)
引 言
目前化石能源仍是人类社会最主要的能源形式,但由于全球储量有限以及对环境造成不可逆转的负面影响,寻求一种兼具环保性、安全性、经济性和可持续发展性的新型能源形式成为全球能源产业变革的热点方向[1]。当前,“降碳减排”是全球能源产业发展的主旋律,我国提出了碳达峰、碳中和目标积极应对全球能源变革。在二次能源载体中,氢能由于来源丰富、燃烧热值高、零碳排放等优点,被认为是一种理想的选择[2]。然而,“氢经济”的推广受到了制氢、储氢、运氢的技术和成本限制[3-4]。
如图1所示,在制氢环节,以煤、石油、天然气为原料的制氢工艺占据着主导地位,2017 年全球氢能产量中工业副产氢和电解水制氢仅占4%,在降碳减排的压力下,亟需一种新型的制氢路线来满足低碳排、高纯度、经济可行的市场需求[5]。在储氢和运氢环节,综合考虑终端用途和输运距离等因素,高压、液化是最常见的储运形式,但其安全性和经济性仍面临着考验[6-8]。在氢的应用端,燃料电池同样面临着“供氢难”的困境,原因是质子交换膜燃料电池(PEMFC)对氢源纯度和杂质成分的要求极为严苛,因为碳氧化物对燃料电池的催化成分具有毒化效应[9-10]。由此来看,传统工业制氢的技术路线已然不能满足当今及未来社会对清洁氢能的广泛需求。
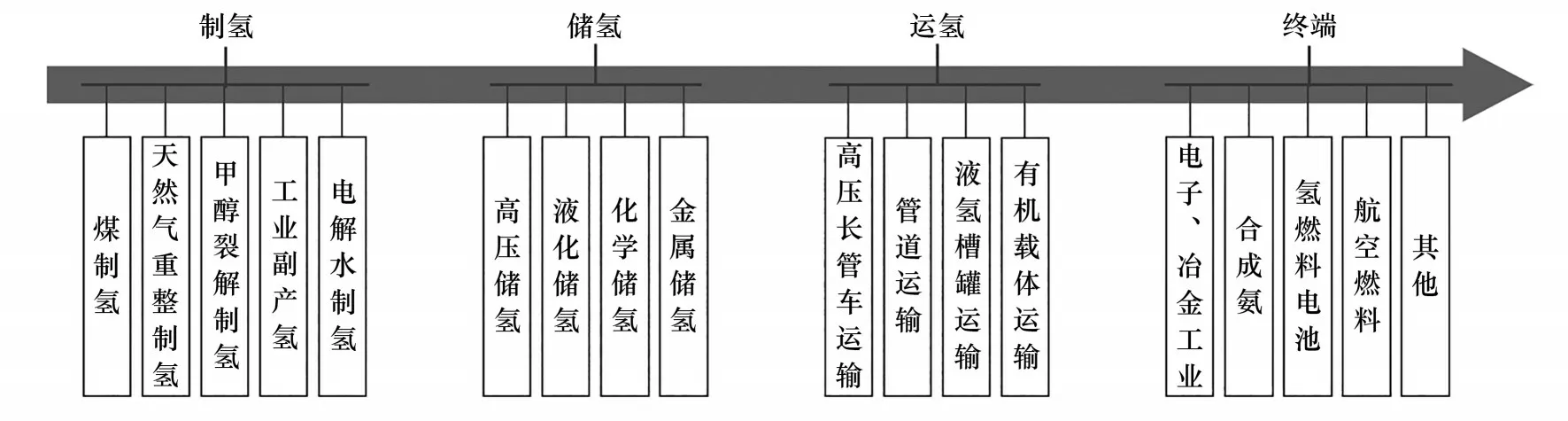
图1 制、储、运氢及终端各环节中的常规技术Fig.1 Conventional technologies in each link of hydrogen production,storage,transportation and terminals
因此,面对氢能利用的尴尬局面,寻求一种低碳环保、经济安全、高效灵活的制氢方案显得尤为重要。近年来,电化学技术在催化化学领域的研究备受关注。在温和条件下,通过电催化分解含氢物质(H2O、尿素、NH3等)制氢的技术路线已成为发展清洁能源的有效途径[11-13]。氨具有高储氢密度(17.6%,质量分数)、运输便利、无碳等优点,在室温(298 K)和较低压力(1~2 MPa)下就能实现液化储运,与甲醇储氢(12.5%,质量分数)、金属储氢等途径相比具有很大潜力,无疑是一种可靠的化学储氢介质[14]。尽管氨具有一定毒性,但在安全浓度水平(25 cm3NH3/m3空气)之下,即可检测到氨的气味。氨的燃爆范围相对较窄(16%~25%),远小于H2的燃爆范围(4%~75%),因此泄漏后几乎不存在可燃风险[15-16]。
目前,全球范围内每年氨的产量超2亿吨,运输及分销网络完善,但主要依赖化石燃料作为工业原料和能源供给,每年向大气中排放约5 亿吨二氧化碳[17-18]。近年来,“绿氨”的概念逐渐引起关注[19]。西门子公司在英国牛津郡开发“绿氨”储能示范项目,由氮气发生器、电解水系统和可再生电力驱动的Haber-Bosch 反应堆组成,展示了以可再生能源生产“绿氨”的现实方案。从这个角度来讲,以氨储氢的技术路线对于我国消纳可再生电力也具有十分重要的意义。不同储氢方式的质量分数和体积密度见图2。
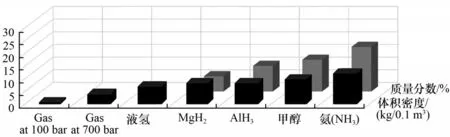
图2 不同储氢方式的质量分数和体积密度[14](1 bar=105 Pa)Fig.2 Mass fraction and bulk density of different hydrogen storage methods[14]
电催化分解氨的过程由两个电极反应组成:阴极析氢反应(hydrogen evolution reaction,HER)和阳极氨氧化反应(ammonia oxidation reaction,AOR)。理论上,电解氨所需的外加电压远小于电解水;具体来讲,电解氨生产1 kg H2需要提供33 MJ的能量,而电解水则要高达180 MJ,但在实际过程中都需要一定的过电位来克服动力学阻力[20-21]。因此,开发高效、稳定的电催化剂在降低反应过电位、提高电流密度方面具有重要意义。碱性水电解质中电催化分解氨示意图见图3。
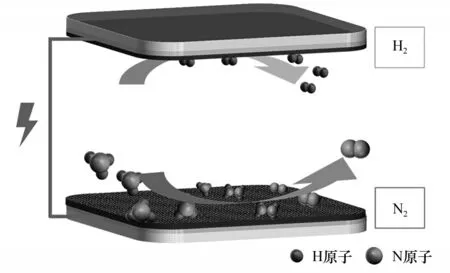
图3 碱性水电解质中电催化分解氨示意图Fig.3 Schematic diagram of electrocatalytic decomposition of ammonia in alkaline water electrolyte
本文以阳极AOR 为关注重点,综述了电催化分解氨的反应机理及催化剂的制备策略和方法,可为开发具有更高活性的AOR 催化剂和发展“以氨储氢”的技术路线提供思路和指导。
1 电解制氢技术
1.1 工业电解水制氢
电解水的概念自发现至今已有200 余年的历史,也是工业电解制氢中最成熟的技术之一。电解单元一般由电源、阳极、阴极和电解质组成[22],如图4所示。直流电源向阴极提供电子以促进质子(H+)的析出,为保持电中性,阴离子通过电解质向阳极转移,使得电子能够形成回路并流入电源正极。为减小传质阻力对电解槽的影响,电解质通常选用传质能力较强的离子溶液。其中,氢氧化钾是最常用的碱性电解质溶液,不选用酸性电解质是考虑到工程中酸腐蚀的弊端。
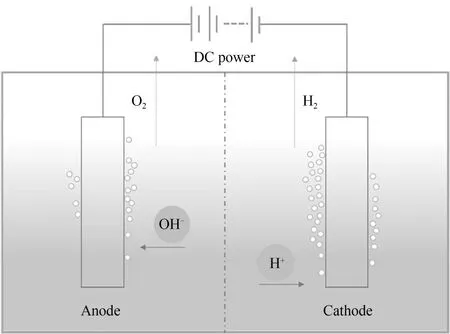
图4 电解水单元示意图Fig.4 Schematic diagram of electrolyzed water unit
水(H2O)能够在自然界长期稳定存在,表明将其分解势必要克服一定的阻力才能实现;对这种阻力进行分析,需要从热力学、动力学以及传质过程等方面加以综合考虑。从热力学的角度,电解水的实现必须克服电化学反应的Gibbs自由能(ΔG),由式(1)可以计算得到电解水的平衡电动势:

式中,n是反应中转移电子量,mol;F是法拉第常数。
在25℃时,电解水反应的Gibbs 自由能变化为+237.2 kJ/mol,转换为平衡电动势E=1.23 V,这是电解水制氢所需的最低能量[23]。从动力学的角度,即使满足平衡电动势这一需求,电极反应仍是缓慢的,为了使反应持续高效地进行,过电位η的产生是实际电解过程中的必然结果[24]。在动力学上,将电解单元中的各种阻力类比为电路中的电阻,可以分为三类:由电路和连接造成的欧姆电阻,由维持电极反应持续进行的极化电阻和由电解质、离子交换膜以及气泡导致的传质电阻。因此,从动力学的角度提高电解效率就需要了解各种阻力的来源并致力于使其降至最低。
根据电解槽技术的差异,电解水技术可以分为碱性电解槽(AEC)、质子交换膜(PEM)电解槽、固体氧化物电解电池(SOEC)和光解水[25]。表1列出了几种电解槽技术的对比。效率最高的PEM 电解槽对组件要求较高,包括昂贵的聚合物膜、贵金属多孔电极。组件和能耗的成本几乎限制了其大规模应用,小型化PEM 水电解器可用于特殊场景,如军事和太空应用。SOEC 和光伏电解的实现需要克服高温、高腐蚀性环境带来的工程性难题和挑战[26]。相比之下,碱性电解水技术成熟、效率尚可,仍是现阶段能够实现大规模制氢的可靠方案。

表1 几种电解槽技术的对比Table 1 Comparison of several electrolytic technologies
目前,全球氢能产量约48%是由天然气重整获得,30%来自石油生产,18%来自煤炭生产,仅有4%的份额来自工业电解水[28]。Kuckshinrichs 等[29]报道了德国、奥地利和西班牙三个国家商业化电解水制氢(AWE)技术的经济性分析。结果表明德国的H2成本约为每千克3.64 欧元,而在奥地利和西班牙成本略高(15%~18%)。此外,Matute 等[30]估计,AWE的H2生产成本约为每千克6 欧元,基于PEM 电解槽的制氢成本为每千克7 欧元。相比之下,天然气重整制氢的成本约为每千克2.08~2.27美元,煤气化制氢的成本在每千克0.36~1.83 美元之间[31]。显而易见,电解水制氢成本远高于传统技术,因此短期内仍无法占据较大的市场份额来取代其他技术。
1.2 电催化氨分解制氢
氨分解制氢可分为热裂解技术[32-34]、等离子体驱动氨裂解技术[35-36]和电催化氨氧化分解技术[20,37-38]。作为合成氨的逆反应,氨热裂解属于吸热反应,在一定条件下氨的转化率受到热力学限制。在450℃时,氨的平衡转化率在99%以上,但由于反应动力学的阻碍限制了反应速率,要实现完全转化只能提高反应温度至700℃及以上[39]。因此,为了提高热催化反应的转换效率,必须由外部持续地提供热量,消耗大量能源且成本较高。
工业上通常采用催化技术来提高生产效率,一般来说,Ru 基催化剂对氨热裂解具有更高的活性,而过渡金属Ni基和Fe 基催化剂更具有经济效益[40]。常见的氨热裂解反应堆包括:适用于固定生产和大型工厂的填充床反应堆,小型反应堆包括微通道、膜反应器[41]等。
近年来,得益于电化学技术的发展,氨电催化氧化技术被更多学者关注。氨电解的理论能耗为2.79 kJ/mol H2,在较低的电压下有望成为一种利用可再生能源生产高纯氢气的替代方案。
1.2.1 氨水体系 在氨水体系中,氨的电氧化反应对溶液pH 较为敏感[42]。吸附在电极表面的氨分子在碱性环境中被OH-氧化或在酸性环境中被HOCl等氧化剂间接氧化为NH+4[20]。在碱性条件下,水解平衡更有利于NH3分子的存在,是AOR 研究中最常用的体系;而酸性条件下,NH3分子转化为NH+4,间接氧化导致其效率较低,电氧化的过程也是缓慢的,同时酸性电解质中的电腐蚀情况更严重[43]。
在碱性电解质中,阳极反应消耗NH3和OH-产生氮气、水和电子[式(2)],阴极反应将水和电子还原为H2和OH-[式(3)]。理论上,该反应在热力学上是有利的,电解电压只需0.06 V,明显低于水自身电氧化所需的1.23 V,所需能耗比电解水要节约95%。然而,AOR 在动力学上却是缓慢的,且阳极氧化反应存在较高的过电位。
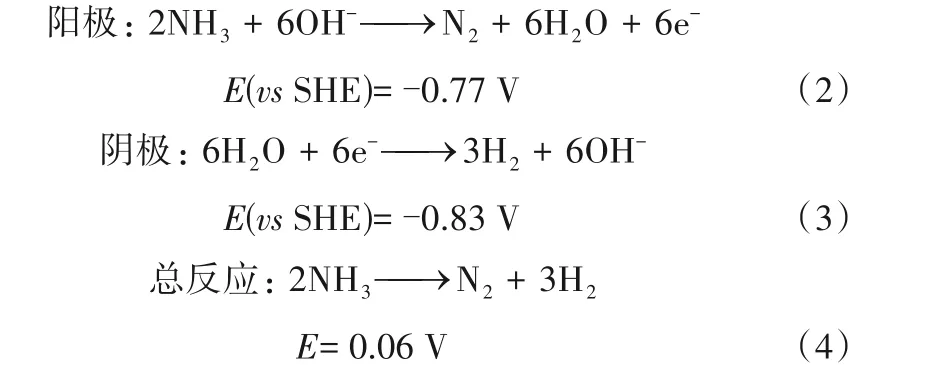
AOR机理是一个极其复杂的过程。近几十年以来的研究中,尽管有很多学者尝试去解释其反应途径,但本质仍未得到充分的验证。1963年,Oswin等[44]针对氨(KOH,aq)在铂电极表面氧化的电化学动力学进行了研究,并将实验得到的Tafel 斜率与基于Temkin 和Langmuir 等温线的理论斜率进行了比较,提出了连续反应的动力学机理。他们认为氨在电极表面连续脱除三个H 原子,然后两个N 原子耦合生成N2(称为“完全脱氢机理”,表示为“O-S机理”)。
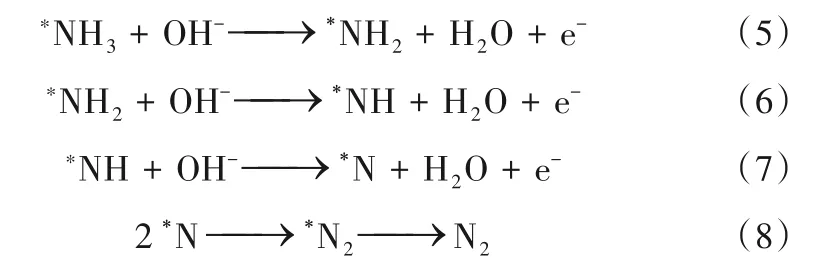
1970 年,Gerischer 等[45]提出了一种完全不同的反应途径,即NH3的氧化是由同时吸附在同一活性位点的OH-引发的,*NH3逐渐脱氢为不同的吸附产物*NHx(x=1 或2),它们重组形成N2Hy(y=2,3 或4),随后脱氢生成N2(称为“部分脱氢机理”,表示为“GM 机理”)。此外,他们还通过平行实验解释了NH3不可能连续脱氢形成N 原子再发生结合,原因是N原子强烈吸附于催化剂表面的活性位点,以至于它们的产生会导致催化电极的不可逆失活。

近年来,随着电化学测试技术和原位表征方法的发展,G-M 机理得到了多种实验方法的验证,包括微分电化学质谱[46](differential electrochemical mass spectrometry,DEMS)、表面增强拉曼光谱[47](surface enhanced Raman spectroscopy,SERS)、红外光谱[48](Fourier transform infrared,FTIR)和旋转圆盘电极[49](rotating ring disk electrode,RRDE)研究。关于碱性水电解质中AOR研究的观点见表2。

表2 关于碱性水电解质中AOR研究的观点Table 2 Views on AOR research in alkaline water electrolyte
1.2.2 液氨体系 液氨电解比氨水电解更具吸引力,原因是液氨的能量密度是饱和氨水溶液的3倍,且不存在碱性环境导致的腐蚀性,有利于运输和存储,在AOR 过程中不会产生氮氧化物这种副产物。为了实现高能量密度的氨电解,研究人员使用碱金属酰胺、铵盐及各种有机溶剂作为电解质来研究液氨电解的机理。从动力学来看,液氨体系下的AOR机理与氨水体系中的G-M 机理不同,这取决于体系所使用的非水电解质类型。
类似于H2O 分子在溶液中能够电离出OH-和H+一样,NH3分子在液氨体系中也能解离为NH+4和NH-2,液氨电解也可以通过以下两个途径开展。
电解质为金属酰胺时
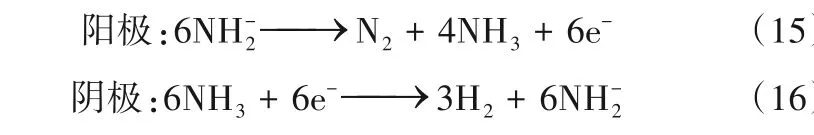
电解质为铵盐时
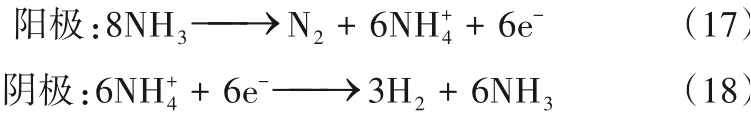
Hanada 等[58]以碱金属酰胺(LiNH2、NaNH2和KNH2)作为电解质直接电解液氨。氨在阴极上还原生成H2,酰胺离子在阳极被氧化为N2。在循环伏安测试中,他们认为三种金属酰胺电解质的性能排序为:LiNH2<NaNH2<KNH2,但并未给出明确的机理解释。在1 mol/L KNH2的电解质体系中,电流效率达到了85%。Goshome等[59]以NH4Cl为电解质进行液氨电解,在2.0 V的电解电压下获得了70 mA/cm2的电流密度,并成功产生20 MPa的高压缩氢气。然而,Pt电极在电解过程中与NH4Cl电解质发生腐蚀反应[式(19)],以至于浸没在电解质中的部分Pt 电极消失。Dong等[60]通过改进Pt电极测试了多种铵盐作为电解质,三元合金Rh-Pt-Ir电极有效降低了阳极电位(最低为0.47 V),并获得了80%以上的电流效率(图5)。
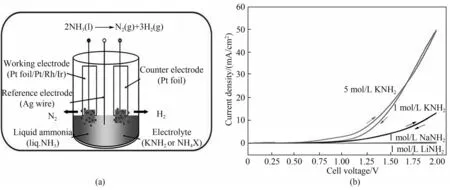
图5 NH4Cl电解质中液氨电解示意图(a);金属酰胺电解质中的液氨循环伏安(CV)曲线(b)[60]Fig.5 Schematic diagram of liquid ammonia electrolysis in NH4Cl electrolyte(a);Cyclic voltammetry(CV)curve of liquid ammonia in metal amide electrolyte(b)[60]
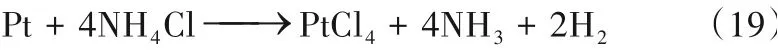
2 AOR电催化剂
催化剂作为电化学过程中至关重要的部分,一直是研究的关键。目前针对阴极析氢反应(HER)的研究相对成熟,阳极AOR 明显受到性能不足、阳极过电位高和副产物(NOx化合物)形成的阻碍。电极表面AOR 机理涉及氨的脱氢和随后中间产物*NHx的聚合脱H 以形成N2。因此,理想的电催化剂应具有足够的脱氢能力,但对N 的吸附作用又不能太强。为使AOR 达到高电流密度、低阳极过电位和优良的稳定性,大多数针对AOR 电催化剂的研究都集中在Pt 这种贵金属上,因为它能够促进NH3脱氢并产生显著的电流密度,吸附态*N 对铂电极的毒害往往导致AOR 反应动力学缓慢和催化剂快速失活。为了解决这一问题,研究人员从调控铂的形貌特征、晶面晶型以及采用合金化(二元或多元)的设计策略进行了探索。另外,开发低成本的非铂AOR催化剂也具有重要的现实意义。
2.1 理论研究
2.1.1 金属-氮结合能对AOR 的影响 电极表面发生AOR 是一个非均相催化的过程,势必要求NH3与金属催化剂之间有一定的吸附作用。目前对AOR机理的研究和氨电解催化性能的优化都是基于Pt催化,原因是Pt 能够遵循萨巴蒂尔规则(Sabatier principle),即有效地将NHx吸附到其表面,同时也利于产物的脱附[36]。氮对各种金属的吸附强度如下:Ru>Rh>Pd>Ir>Pt>>Au,Ag,Cu[61]。就Ru、Rh 和Pd 而言,尽管它们对N具有很高的吸附能力,使氨的氧化电位比Pt 低得多,但由于NHx的快速去质子化和随之产生的*N 对表面的不可逆吸附,而导致更快的催化剂失活。相反,Au、Ag 和Cu 具有较低的N 吸附能力,实验证明在AOR中并不活跃。
Ir、Pt 介于两种极端情况之间,具有一定的吸附强度来确保在一定电位下可以实现AOR。Ir氧化氨的起始电位低于Pt,通常用Pt、Ir 两种金属组合作AOR 催化剂,与单金属Pt 电极相比,双金属电极会产生更高的氮选择性和较低的中毒效应,并且发现这种组合方式能够显著降低阳极过电位,然而电极表面的电流密度也随之降低[62]。
2.1.2 第一性原理与密度泛函数计算 第一性原理(first principle)是运用量子力学的手段,根据原子核与电子之间的相互作用及其基本运动规律,根据实际需求来求解薛定谔方程的一种算法;密度泛函数(density functional theory,DFT)计算是基于复杂的多电子波函数进行的,现已广泛应用于预测各种分子、纳米粒子和散装材料的基态结构。在催化化学领域,理论计算也往往被用于预测、设计和评价催化剂。
Skachkov 等[63]通过组合第一原理分子动力学和密度泛函理论对Pt(100)晶面的AOR 机制进行了全面理论研究。研究表明,AOR 在中电位区(>0.5 V)遵循O-S机制,在低电位区(<0.5 V)遵循M-G机制。在NaOH 碱性溶液中Pt(100)晶面吸附NH3的示意图见图6。
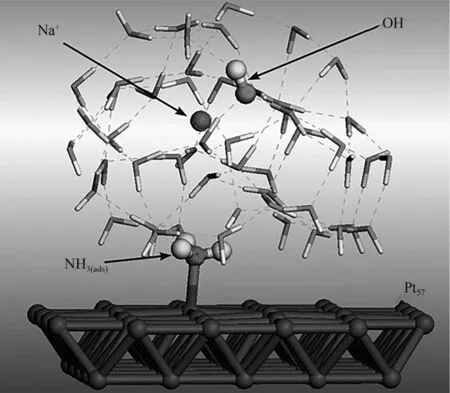
图6 在NaOH 碱性溶液中Pt(100)晶面吸附NH3的示意图[63]Fig.6 Schematic diagram of NH3adsorption on Pt(100)crystal plane in NaOH alkaline solution[63]
Herron 等[64]研究了氨在Au、Ag、Cu、Pd、Pt、Ni、Ir、Co、Rh、Ru、Os 和Re 模型表面的电催化过程,结果支持了G-M 机理,即形成N2是由NHx的二聚脱氢而不是*N 原子的耦合,并预测了Pt 是活性最高的金属,其次是Ir和Cu。然而,实验证明Cu对AOR 无活性,认为这可能是在分析中直接比较N—N 键形成步骤的活化能与质子-电子转移步骤的吉布斯自由能而导致的,除了热力学的能量“屏障”,很可能还存在动力学因素阻碍Cu 产生活性。Estejab 等[65]对Pt3-xIrx(x=0~3)合金催化剂上的AOR 进行了密度泛函理论计算,发现在Pt基催化剂上AOR机制包括形成N 的二聚物而后脱氢成N2,Ir 基催化剂上则以氨连续脱氢成*N 原子而后形成N2,计算结果还表明Ir上AOR 的起始电位低于Pt,电氧化反应从Ir 的低电位开始。两种AOR 机理下金属-N 结合能与活性的关系,Pt和Pt-Ir催化下的AOR机制见图7。
2.2 AOR电催化剂
2.2.1 贵金属催化剂 贵金属Pt 是催化AOR 的最佳选择[64]。目前对AOR机理的研究及电催化性能的优化大多都是基于Pt催化来开展的。根据文献报道,在使用Pt 电极时,AOR 几乎只在Pt(100)晶面发生,而Pt(110)、(111)晶面少有活性,从侧面佐证了AOR是对结构十分敏感的反应[63,66]。Katsounaros等[67]在G-M机理的支持下研究了Pt(100)晶面上氨的氧化机理,认为通过N—N 键的耦合形成N2分子是不太可能的,因为这在化学反应的能量变化上是不利的。N—N 键的耦合会被具有强吸附能力的*N 和*NO 所抑制,而这些产物是由*NH 进一步氧化形成。因此,为提高Pt 基催化剂的活性和稳定性,应加速二氮的耦合而抑制*NH 的进一步氧化,使催化反应向着有利于生成N2的方向进行。
贵金属Ir 对AOR 也具有催化活性,因其较低的起始电位和适中的N 结合能力,也被认为是AOR 潜在的催化剂。Zhang 等[68]在Ir(100)晶面上进行的密度泛函数理论计算结果证实了这一点,他们得到了NHx(x=1,2,3)在Ir 表面的吸附能量以及每个脱氢过程中N—H 键断裂的能量屏障,发现NH2是最稳定的表面吸附物种,并在反应中抑制NH3的第一次脱氢,成为决速步骤。在Botte等[69-71]的系列研究中,以降低AOR 过电位及提高电流密度为优化目标,认为在Pt中添加Ir能够提高AOR 的催化效果,此外他们还尝试添加Ru、Rh 来进一步优化。Endo 等[62]、Lomocso 等[72]也报道了Pt-Ir 在氨氧化进程中产生更高的活性(优于Pt),这可能要归功于双金属之间的电子效应和协同催化的结果。
2.2.2 非贵金属催化剂 为实现AOR 在实际生产中的应用,研究必须克服贵金属催化剂的高成本、失活停用等问题。在非贵金属催化领域,镍、铜、钴、铁等金属材料被用来研究以替代贵金属基催化剂。其中,Ni 是AOR 中研究最广泛的催化剂之一。纯Ni 电极对AOR 没有活性[73],Kapałka 等[42]将Ni 电极在碱化电解液中进行循环伏安(CV)扫描,使金属Ni 转变为Ni(OH)2之后获得了AOR 活性。其原因是Ni(OH)2在碱性溶液中可进一步地氧化为高价态的活性物质NiOOH,即发生Ni(OH)2—NiOOH 这一可逆的氧化还原反应,而NiOOH 具有很高的催化氧化活性,能够将氨氧化成N2。关于贵金属催化AOR 的研究见表3。

表3 关于贵金属催化AOR的研究Table 3 Research on AOR catalyzed by noble metals
Allagui等[83-84]以Pd为成核元素研究了多元醇法合成的纳米Ni 催化剂,纳米Ni 颗粒在50~100 nm 之间。在碱性溶液中进行CV 扫描以激活催化剂形成Ni(OH)2层并开展电化学测试。结果发现,由于NH3和OH-之间的竞争关系以及Ni和N原子之间的强结合能力,低电位下不会发生H2O的电氧化。此外,在NiOOH 氧化NH3的同时,自身也会被还原为Ni(OH)2。
Ni 和Cu 的协同效应也被用于研究催化AOR。Xu 等[85]通过电化学沉积在碳纸上制备了NiCu 催化剂,颗粒大小为100 nm 左右。在氨电解电池(AEC)中,催化剂表面被氧化为金属氧化物和氢氧化物,在高pH 环境和高电解电压(>1.0 V)时,产物形成了NO-3,相比于对照实验的Ni/C 和Cu/C 电极,NiCu/C电极的AOR 电流密度提高了10 倍以上,达到了8.5 mA/cm2。Cu 的存在不仅提高了Ni 的催化活性,还改善了电极的稳定性。尽管如此,Ni 基催化剂的起始电位仍高于Pt 基催化剂,且氧化电流低。在高电位区间容易与H2O氧化产生竞争。
2.3 阳极电极的制备
催化剂的制备方法在一定程度上决定了它的催化性能。为了改善电催化剂的性能,电化学沉积、多元醇、共沉淀、水热合成等方法已被广泛应用。催化剂的获得是制备催化电极的第一步,本文根据金属催化剂的制备方法,总结了几种常用的阳极电极制备方法。
2.3.1 电化学沉积法 电化学沉积法是一种高效、精确、可扩展的催化剂制备方法。一般在三电极电镀槽中进行,外加电场可以将分布均匀的催化剂颗粒直接沉积到催化剂基底上。众多文献都采用电化学沉积的方法来制备AOR 催化剂,并对沉积条件进行探索和优化,包括沉积电压、电镀液酸碱度、操作温度等。Gwak 等[89]研究了不同沉积条件下制备Pt 催化剂并使用该催化剂在碱性介质中进行氨电解。在不同的沉积电压下,制备得到树突状(-0.2 V)、金字塔状(-0.1 和0 V)、花椰菜状(+0.1 V)和光滑球状(+0.2 V)等不同形貌的Pt 催化剂。在电解实验中,他们发现强碱性(1 mol/L KOH)和高浓度(1 mol/L NH4OH)能够促进氨的电解行为,高电解电压促使金属氮化物的形成并导致催化剂失活。Gwak 等的研究成果见图8。Zhou 等[81]开发了一种热退火-电位动力学电位策略,以制备混合催化剂,其中Pt 颗粒通过电沉积沉积在N 掺杂石墨烯(Pt-NG)上。

图8 不同电压下在碳纸上电沉积Pt的SEM图像[(a)~(e)]和10 mmol/L H2PtCl6+0.5 mol/L HClO4溶液中恒电位沉积期间的I-T曲线(f)[89]Fig.8 SEM images[(a)—(e)]of electrodeposited Pt on carbon paper and current-time profile during potentiostatic deposition in 10 mmol/L H2PtCl6+0.5 mol/L HClO4 solution at various voltages(vs Ag/AgCl)(f)[89]

表4 Ni基催化剂催化AOR的相关研究Table 4 Research on Ni-based catalysts catalyzing AOR
Xu 等[85]报道了在碳纸基底上电化学沉积NiCu双金属催化剂并应用于低电压下处理含氨废水。扫描电镜图显示金属颗粒均匀地沉积在碳纤维上并伴有约100 nm 大小的团聚现象,Ni 和Cu 的分布并不均匀,产生了富镍和富铜区域,XRD 衍射图像也证实了两种金属独立存在而不是以合金的形式被沉积(图9)。Liu 等[80]报道了另一种结合电沉积和电镀锌置换的合成方法:首先,花状镍颗粒被电沉积在导电基板上,通过替换部分镍颗粒加载Pt。这种方法保持了花状形态和纳米孔以及小尺寸的Pt颗粒,导致质量活性为75.32 mA/mg(比商用Pt/C高2倍)。

图9 NiCu/CP电极在氨电解测试之前(a)和之后(b)的SEM图像;NiCu 催化剂的STEM 图像(c);NiCu 催化剂中的Cu、Ni分布(d)[85]Fig.9 SEM images of the NiCu/CP electrode before(a)and after(b)ammonia electrolysis tests;STEM images of NiCu catalyst(c);Cu,Ni distribution in the NiCu catalyst(d)[85]
2.3.2 超声-涂覆法 在实现催化剂颗粒纳米化、可控化的研究中,多元醇法、共沉淀法、水热合成法是常见的合成手段。这些方法合成的催化剂颗粒并不能直接用于电化学实验或催化反应,需要将其负载于导电基底上制备阳极电极。超声-涂覆法是指将催化剂浆料(由催化剂细粉、聚四氟乙烯乳液或Nafion 溶液与醇类溶液混合而成)在超声浴中分散均匀,涂覆在目标基底材料上烘干即得到催化电极。在进行AOR 电化学测试之前,需将阳极电极在碱性溶液中进行CV 扫描以激活工作电极。
Song 等[82]报道了碳材料基底负载Pt、Ir 和PtIr(1∶1)合金催化剂的AOR 动力学研究。在60℃下,该催化剂在恒氨浓度的1 mol/L KOH 溶液中进行电化学测试。与Pt相比,Ir随温度的升高,阳极过电位显著下降的同时峰值电流也降低,PtIr合金结合了Ir和Pt 的优点,表现出更宽的电解电位窗口和更高的峰值电流。
Wang 等[90]在超声和搅拌的操作下还原Ir3+、Co2+并合成碳基底(XC-72R)负载的It-Co/C 纳米复合催化剂(图10),以涂覆该催化剂的玻碳电极(GCE)作为工作电极,研究其在0.4 mol/L NaClO4溶液中对AOR的催化性能,还提出了AOR催化剂在氨检测应用中的方案,论证其比商业PtRu/C 具有更好的灵敏度和响应速度。
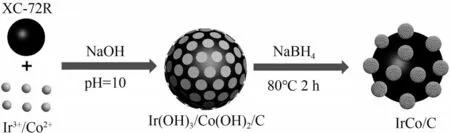
图10 Ir-Co/C催化剂合成过程示意图[90]Fig.10 Schematic representation of the synthetic procedure of Ir-Co/C[90]
Li 等[91]通过超声辅助在多孔二氧化硅(SiO2)和功能化碳纳米管(CNT-COOH)组成的复合基底上成功负载了三元PtIrNi合金纳米颗粒(图11),并在碱性介质中对AOR 催化性能进行了测试。室温下,阳极过电位(0.40 V,vsRHE)低于商用PtIr/C(0.43 V,vsRHE)。增加NH3浓度和操作温度可以显著降低阳极过电位并提高峰值电流密度,在80℃时阳极过电位下降为0.32 V(vsRHE)。
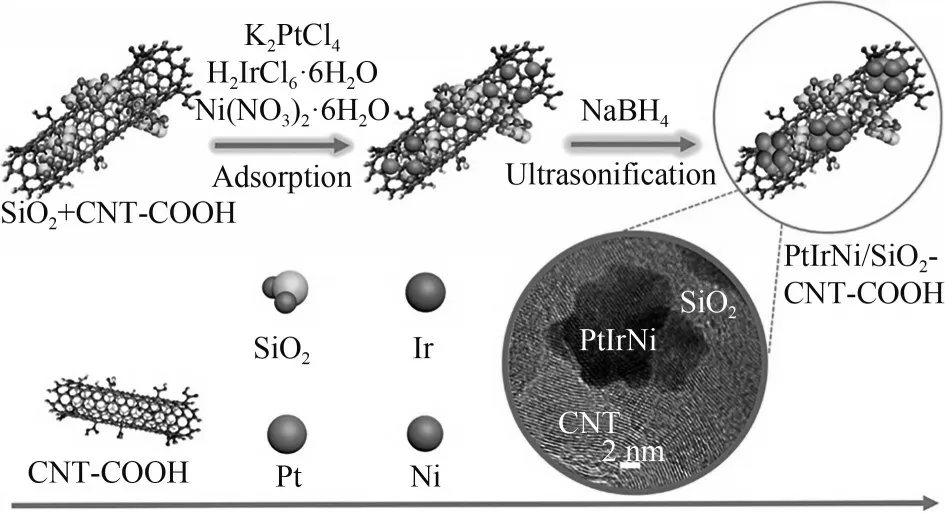
图11 SiO2和CNT-COOH负载PtIrNi三元合金示意图[91]Fig.11 Schematic illustration for the synthesis of SiO2 and CNT-COOH-supported PtIrNi ternary alloys[91]
2.4 催化剂的基底材料
催化剂载体在调控电子结构、增强物质传输和增加活性位点等方面发挥着重要的作用,选择合适的载体材料对提升AOR 性能也至关重要。碳材料因其机械稳定性和良好的导电性能,是各项研究中最常用的载体之一,包括铂碳(XC-72R)、碳纳米纤维(CNF)、石墨碳、碳纳米管(CNTs)等。碳载体负载活性物质作为催化电极,既能保证良好的电子传导效应,高比表面积,还能够提供丰富的催化活性位点,二维、三维碳材料(如石墨烯、碳纳米管)负载的催化剂往往表现出更高的催化活性和稳定性。
Zhang 等[92]认为由于电子效应的作用,AOR 中间体的吸附作用减弱并促进了化学键的断裂,从而提高了贵金属催化剂的反应活性。Zhou等[93]在N掺杂的还原氧化石墨烯(N-rGO)上负载了贵金属Pt和Ir,并在碱性介质中研究了氨电氧化反应。与商业Pt/C(20%,质量分数)相比,Pt-Ir/N-rGO 获得了更高的电流密度和更低的中毒效应,表明Pt、Ir 和N-rGO 底物之间存在协同效应,有利于电催化活性。在另外一份报告中[94],他们成功制备了一种负载Pt 纳米颗粒的三维多孔N 掺杂石墨烯气凝胶(NGA)用于氨电池(ammonia electrolysis cell,AEC),由于均匀分散的Pt 纳米簇状形态、优良的导电性、3D多孔NGA网络以及石墨烯框架中的N掺杂结构,获得了1.77 mA/mg(Pt)的质量活度和0.64 mA/cm2(ECSA)的比活度。
此外,SiO2基底、多孔Al2O3也用于催化剂的负载和研究。就文献报道来看,目前更多的研究仍致力于探究AOR 的催化机理并提升催化剂的活性,针对基底材料的研究和综述并不多。
3 总结及展望
氨作为一种无碳、高储氢密度的液相介质,在温和条件下通过电催化分解氨制氢,具有广泛的研究价值和应用前景。近几年,以氨储氢的技术路线在国内外成为研究热点,电催化氨分解在富氨废水处理[73,95]、现场制氢[96]、电化学传感[97-98]及燃料电池[99-100]等领域的应用也广受关注。
然而,AOR 电催化剂的性能不佳和贵金属的高负载量是限制电催化氧化氨广泛应用的技术阻碍。贵金属催化剂具有得天独厚的低电位性能和高电流密度,但仍存在催化剂中毒和高成本的问题;非贵金属催化剂在AOR 中存在较高的阳极过电位,这就会导致氨氧化与水氧化产生竞争并生成副产物NOx(x=1,2),同时AOR 的电流密度仍有待提高。相比氨水体系,液氨电解的优势更加明显。液氨具有高能量密度,体系中不存在与水氧化的竞争和酸碱腐蚀的问题。但受限于低温环境,对液氨体系的AOR 研究相对较少,针对反应机理的探究和催化剂的开发仍有待深入。
因此,开发催化性能更好、贵金属负载量更低或非贵金属催化剂成为突破上述技术难点的研究方向。不少研究者尝试从纳米化、合金化、形貌调控、电子调控等方面去优化AOR 电催化性能,主要总结为以下几点:(1)催化粒子纳米化。通过降低催化颗粒的尺寸,达到增加催化表面积和活性位点的目的。(2)催化金属合金化。金属合金具有协同作用,可以起到调节催化性能的重要作用。(3)形貌调控。通过改进制备方法、添加助剂等手段,对有利于催化反应的金属晶型、形貌特征进行可控生成,从而提高催化性能。(4)电子调控。载体与催化颗粒之间的相互作用可以调节金属的电子结构,对活性位点的分布也具有重要作用。
针对上述研究思路,本文提出以下研究建议:(1)以理论计算(如DFT)与实验探究相结合的方法,借助人工智能或神经网络等手段研究AOR 机理及活性位点,并指导AOR 电催化剂的设计与优化;(2)金属有机框架(MOFs)材料作为一类由过渡金属节点与有机配体相构建的新型材料,具有微观形貌可控、高比表面积等特征,在电催化领域广受关注,可以尝试合成非贵金属基MOFs 催化剂应用于电催化氨,关注AOR 应用场景并对工业化放大工艺进行探索,如现场制氢技术、工业废水治理技术、电化学传感器等;(3)基于全生命周期对氨这一氢载体进行全面评估,包括生产技术、运输成本、应用价值乃至技术因素等,使用量化的手段对“氨经济”做出科学、全面的评估。
猜你喜欢
物理化学学报(2024年9期)2024-09-27 00:00:00
长江蔬菜(2021年22期)2022-01-12 03:25:36
上海建材(2020年12期)2020-04-13 05:57:52
长江蔬菜(2018年22期)2018-12-25 12:37:22
长江蔬菜(2018年6期)2018-05-08 07:45:10
中国有色金属学报(2018年2期)2018-03-26 07:58:37
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
当代化工研究(2016年5期)2016-03-20 16:21:32
电源技术(2015年11期)2015-08-22 08:50:26
低温与特气(2014年4期)2014-03-20 13:36:50
