
致病性钩端螺旋体的多位点序列分型研究
2022-03-18 10:07杜宗利辛晓芳徐颖华
中国人兽共患病学报 2022年2期
李 喆,张 影,杜宗利,辛晓芳,叶 强,徐颖华
钩端螺旋体病(简称钩体病)是由致病性钩端螺旋体(简称钩体)引起的人兽共患传染病[1]。尽管随着经济的发展和卫生条件改善,在过去几十年世界各国钩体病的发病率大幅降低,但据WHO统计,每年仍有约200万钩体感染病例,一些地区甚至还发生暴发流行。因此,钩体病仍是当前全球的重要公共卫生问题[1-2]。
病原体的监测和溯源是预防和控制传染性疾病的最关键步骤之一。国际和国内对钩体菌株的分类从最早血清学分类法,到各型不同原理的分子分型方法[3-5]。随着分子生物学的发展,自20世纪90年代起,研究人员就开始应用16SRNA基因测序方法对钩体进行分子分型,并将致病性钩体中包括问号(Leptospirainterrogans)、波氏(L.borgpetersenii)、卫氏(L.weilii)等10个基因种[6-7]。此外,Ahmed等[8]于2006年首次将基于多种管家基因的多位点序列分型法(Multi-Locus Sequence Typing, MLST)技术用于钩体的分子分型,由于该方法简单、快捷、结果易于比较,被广泛用于不同致病性基因型的分子分型、进化溯源和种群结构等研究[9-10]。而目前对全球致病钩体菌株进行系统性MLST分析的研究较少。此外,中国致病性钩体菌株MLST研究多用于黄疸出血群的型群结构分型[6,9],而其他血清群致病性钩体分型和遗传多样性的研究报道也较少。因此,在本研究应用16SRNA基因测序和MLST分析方法对不同来源、不同时期的21种不同血清群的国内钩体分离菌株与国际参考菌株进行分析,并与国内7株疫苗株结果平行比较[4]。同时基于PubMLST数据库和已有的文献报道[4,6,9],共收集来自世界7大洲不同国家和不同时期1 238株致病性钩体基因种和MLST数据(含236株中国致病性钩体MLST数据)。探讨分析致病性钩体MLST基因多态性,以其系统性了解中国境内和全球致病钩体菌株种群结构和遗传进化关系。
1 材料与方法
1.1 菌株 致病性钩体国际和国内参考菌株均来自中国食品药品检定研究院。
1.2 主要试剂及仪器 DNA提取试剂盒购自美国Promega公司;Ex Taq DNA聚合酶和DNA marker DL1000购自大连TaKaRa公司;其他化学试剂均为分析纯;2400型基因扩增仪为美国Applied Biosystems公司产品。
1.3 细菌培养和DNA提取 将钩体菌株接种于含10%兔血清的磷酸盐培养基中,28 ℃培养14 d。离心收集菌体,按照说明书进行菌体基因组的提取,基因组DNA样品放置-20 ℃保存备用。
1.4 16SrRNA基因序列分析 以提取的钩体菌株基因组DNA为模板,通过扩增和测序近全长16SrRNA基因(扩增引物见表1),进行钩体基因型的鉴定。将测序结果与参考序列(GenBank: AY631894.1)进行比对,参考文献[7], 根据位点差异判定菌株基于16SrRNA序列的基因型。
1.5 MLST分析 参考文献[10],选择7个国际通用的MLST位点,已提取基因组DNA为模板,应用7种不同管家基因引物(见表1)进行MLST不同位点的扩增及测序。将管家基因测序结果上传PubMLST数据库(https://pubmlst.org/),获得待检测钩体菌株MLST型(ST)。

表1 本研究所用引物序列
1.6 MLST数据收集和系统发育分析 从PubMLST数据库(https://pubmlst.org/)下载已明确MLST型(scheme 1)数据及其菌株信息,同时从国内已发表的相关文献[4,6,9]收集中明确MLST型(scheme 1)的钩体菌株信息。使用Bionumerics软件v7.5 (Applied-Maths)对本研究所有钩体菌株MLST信息进行系统发育分析,并构建最小生成树,并以此探讨致病性钩体种群结构与菌株分离宿主来源、区域等流行病学信息的相关性。
2 结 果
2.1 致病性钩体16SrRNA基因序列分析 16SrRNA基因测序分析结果显示(图1),5株钩体国际参考菌株为问号基因种,其他2株国际菌株为L.kirschneri基因种;而在47株钩体中国分离株中,分别包含28株(59.6%)问号基因种、8株(17.0%)波氏基因种、6株(12.8%)卫氏基因种、4株(8.5%)L.alexanderi基因种和1株(2.1%)L.kirschneri基因种。进一步分析发现6株卫氏基因种菌株除了来源人以外,还来自3种不同动物宿主(图1)。不同菌种的血清群与其基因种没有明显的相关性。
2.2 致病性钩体MLST分型 通过对54株钩体菌株进行7个管家基因位点的扩增、测序及与PubMLST数据库参考序列进行比对,确定了每个菌株每个管家基因位点的基因型,通过组合不同位点基因型最终确定了每个菌株的MLST型(图1)。7株国际参考菌株呈现7种不同ST型。
在47株中国钩体分离株中,发现15种ST型以及24种尚未报道的新ST型(图1)。其中56620、56666、56679和56660菌株均为ST1型,与疫苗株黄疸出血群赖株(56001)相同,这5种ST1型菌株均为问号基因种,56620和56679均为黄疸出血群。此外,还发现56003和56626菌株的ST型与疫苗株犬群611菌株相同,均为ST37型(图1)。
上述菌株MLST分析结果的UPGMA发现,属于问号、波氏和卫氏基因种菌株均各自形成单独聚类分支。而4株L.alexanderi基因种聚类分支中包含一株L.kirschneri基因种菌株。在问号基因种中,7株不同ST的疫苗株也位于不同亚分支中(图1)。
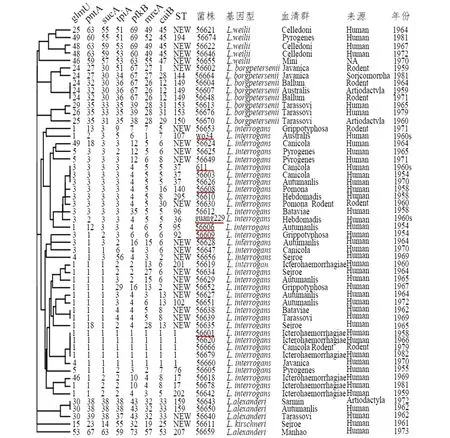
红色下划线标注的为中国钩体疫苗生产菌株。
2.3 致病性钩体基因种与MLST型的多态性分析 共收集(数据库下载、文献引用和本研究结果)1 238株钩体MLST数据。对每一个菌株来源、分离时间、基因种和MLST型进行汇总分析(表2),世界范围内近百年分离的1 238株钩体菌株共分为8个基因型,而中国236株钩体菌株共分为5个基因型,不同国家流行菌株的基因种和ST型不尽相同(表2)。在钩体流行疫情比较严重的亚太地区和南美区域,在过去60多年中,分离钩体菌株基因种主要为问号基因种。例如1962-2016年巴西分离的113株钩体菌株中,83株为问号基因种,相类似,在泰国分离的125株菌株中,92%(115株)的菌株基因种同为问号基因种。而在疫情较轻的非洲国家刚果分离的16株菌株中,12株为L.kirschneri基因种和4株为波氏基因种(表2)。
在不同国家分离菌株中所含有ST型数量也不同,导致其多态性指数也不同,例如在中国和泰国,分别在236株和125株分离菌株发现82种和51种ST型,多态性指数分别为0.35和0.41,而在马来西亚,31株菌株中含有30种ST型,多态性指数为0.97。在澳大利亚分离的16株菌株中发现12株ST型,多态性指数为0.75(表2)。
2.4 致病性钩体种群结构分析 基于MLST系统发育分析结果表明,世界范围内1 238株钩体菌株包含16个主要的进化簇(不少于10菌株/簇),共计669个菌株(54.0%),主要包括ST17、ST34、ST149、ST1和ST37等(表3),其中ST17和ST149遍布全球多个洲分布。236个中国钩体菌株包含4个主要的进化簇(不少于10菌株/簇),分别为ST1、ST128、ST17、ST143,共计120个菌株(50.9%)(表2),ST1与ST128仅有ptnA管家基因不同,其余6个管家基因型均为1型。

表2 不同国家钩体分离菌株的基因种与MLST型

表3 主要流行MLST型钩体菌株的分布
此外,MLST分型与16SrRNA序列分析结果具有较好的一致性,而与血清群分型结果一致性较差,相同的血清群菌株内ST型众多。相同ST型菌株具有不同血清群。进化树分析也显示致病性钩体菌株在不同宿主物种广泛地交叉感染,不同国家间存在广泛传播。分离时间与钩体种群结构分布并不具有明显的相关性(图2)。
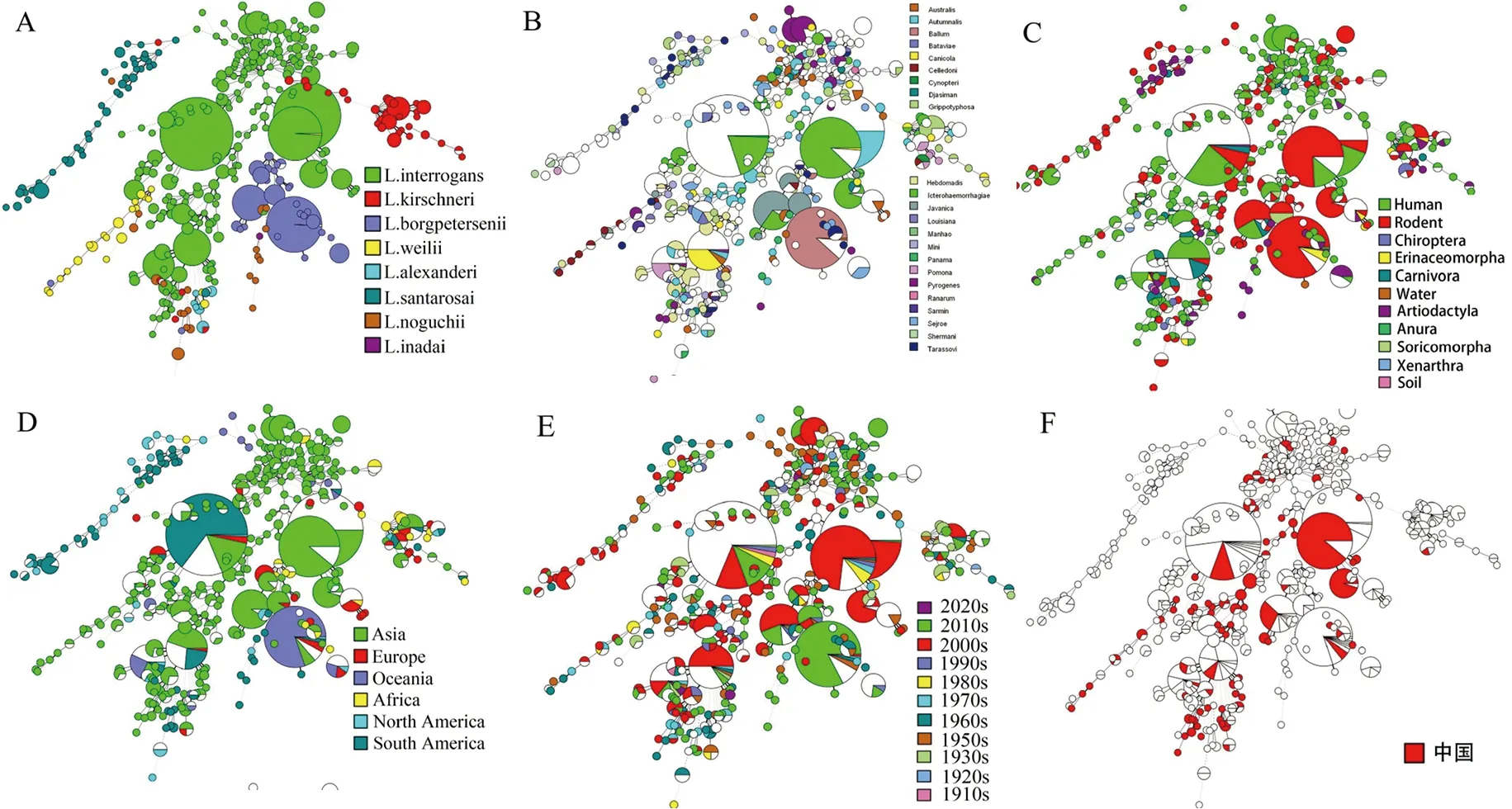
每个圆代表一个MLST型(ST),圆的大小代表一个给定ST的菌株数量。A为MLST与钩体基因型的相关性,不同颜色代表了钩体菌株基于16S RNA测序的不同基因型;B为MLST与钩体血清群的相关性,不同颜色代表了不同的血清群;C为MLST与钩体菌株宿主/媒介的相关性,不同颜色代表了不同的宿主/媒介;D为MLST与钩体分离地区的相关性,不同颜色代表了不同的分离地区;E为MLST与钩体分离时间的相关性,不同颜色代表了不同的分离时间段;F为中国钩体菌株MLST型别的分布。
3 讨 论
目前,全球已发现了近30个血清群300多个型不同血清型钩体,至今仍不断有新的血清型钩体报道[4-5]。在中国,已发现的致病性钩体有18个血清群76个血清型,但目前主要流行血清群仍是黄疸出血群[5-6]。但由于血清学方法检测时需要一系列的标准菌株和标准分群(型)血清,操作繁琐,费时费力,仅在部分钩体参考实验室方可开展,从而限制钩体的分型分析广泛应用。本研究应用简单快捷的16SrRNA和MLST分析方法对不同来源、不同时期的多种不同血清群国内分离菌株和国际菌株进行分析,本研究结果进一步证实16SrRNA和MLST方法不仅可以用于钩体的分子分型研究,还同样适用于不同基因型菌株的进化关系。
我们分析结果发现在47株国内分离的钩体菌株中,多数(59.6%)菌株均为问号基因种,进一步支持在中国过去60多年中分离钩体菌种主要以问号基因种为流行趋势现状[6,11]。同时,还发现在波氏基因种的菌株来自多种动物宿主。以前研究结果证实波氏致病性基因种的基因组含有大量的假基因,较问号等基因种菌株基因组小400~800 kb[11-12],提示其经历不同进化历程,丧失近祖先在普通环境生存的能力,仅能在动物宿主体内寄生,这些分析结果也解释了为什么波氏基因种钩体可在多种动物宿主分离的原因之一。
MLST分析结果证实47株国内分离菌株存在39种ST型,其中包括24种新型ST型,也进一步表明中国钩体菌株资源丰富、菌型复杂,种群结构独特[6,11,13]。此外,尽管当前国内钩体疫苗生产用菌种是依据主要流行血清群的选择代表性菌株,通过MLST系统发育分析发现7株疫苗株位于不同进化亚分支上,从分子水平层面分析,作为疫苗株也具有一定代表性。
由于钩体病属于人兽共患的自然疫源性疾病。其动物宿主的种类繁多,当地区域性的气候、环境变迁可能会影响致病性钩体菌株的遗传变异和进化,表明钩体菌株的基因型与地域具有一定相关性[14-15]。本研究MLST分析也证实不同国家流行不同的ST型,例如钩体疫情严重的泰国和巴西主要流行型分别为ST34和S17,而在中国主要为ST1。由于分型原理不同,血清群与MLST分型没有明显的相关性,相同的血清群菌株具有不同的ST型,反之亦然。中国疾控中心传染病预防控制所收集过去50多年国内5个不同省市的120株黄疸出血群钩体菌株进行MLST多态性分析,证实黄疸出血群群内分化明显,呈现17种不同ST基因型;尽管其中69株钩体为ST1型,但随着时间推移,菌株基因多态性呈现逐渐增多,国际主要流行ST17型占比呈上升趋势[6]。研究人员应用MLST方法对近些年泰国持续暴发钩体疫情进行分子流行病回顾性研究证实,尽管在泰国长期以来主要流行钩体菌株的血清型并没有明显改变,但在暴发区域感染病人分离钩体菌株及同时期其他泰国省市的人和动物宿主分离的MLST型主要为ST34型(分别占比76%、71%和88%),然而在泰国以前分离菌株中并没有发现ST34型,表明流行菌株管家基因发生变异,呈克隆性扩张,形成主要新ST34基因型,在钩体动物宿主中具有较强的传播能力,从而引起钩体病的流行暴发[16]。因此,这些研究结果提示我们在当前流行趋势下,不仅要对钩体流行菌株的血清群(型)进行监控,同时也应加强钩体分子流行病学的研究,掌握钩体主要流行菌株基因型的动态变化,从而为钩体流行病暴发以及突发公共卫生事件菌株溯源和疾病控制提供技术支撑。
利益冲突:无
引用本文格式:李 喆,张 影,杜宗利,等. 致病性钩端螺旋体的多位点序列分型研究[J]. 中国人兽共患病学报,2022,38(2):95-101. DOI:10.3969/j.issn.1002-2694.2022.00.024
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
世界科学技术-中医药现代化(2022年3期)2022-08-22
中国土壤与肥料(2021年5期)2021-12-02
昆明医科大学学报(2021年5期)2021-07-22
中国动物保健(2015年4期)2015-10-21
小天使·五年级语数英综合(2015年7期)2015-07-06
智能制造(2015年4期)2015-05-12
小学生·多元智能大王(2014年11期)2014-11-21
小朋友·快乐手工(2009年5期)2009-06-11
