
Integration of metabolomics and transcriptomics revealed the biosynthetic mechanism of anti-parasitic compounds in Salinivibrio proteolyticus strain YCSC6*
2022-03-05 09:36GuangxunDULingyunQUPingGAOWenhaoHUANGChenghuaLIDewenDING
Guangxun DU , Lingyun QU , , Ping GAO , Wenhao HUANG , Chenghua LI ,Dewen DING
1 School of Marine Sciences, Ningbo University, Ningbo 315832, China
2 Laboratory for Marine Fisheries Science and Food Production Processes, Pilot National Laboratory for Marine Science and Technology (Qingdao), Qingdao 266237, China
3 Key Laboratory of Marine Eco-Environmental Science and Technology, First Institute of Oceanography, Ministry of Natural Resources (MNR), Qingdao 266061, China
Abstract The fermentation broth of Salinivibrio proteolyticus strain YCSC6 shows potent anti-parasitic activity against Uronema marinum, with activity varying in each fermentation stage. To investigate the biosynthetic mechanism of anti-parasitic compounds in strain YCSC6, a comprehensive analysis of metabolomics and transcriptomics over four diff erent time points (12, 24, 48, and 72 h) was performed.Metabolomics detected 17 943 metabolites with 1 129 known metabolites. A trend analysis of the known metabolites showed that 575 metabolites, including 69 polyketides, were continuously enhanced, being the potential source of anti-parasitic agents. In addition, 941 genes mapped to the same pathways of these metabolites, were screened through the association analysis of metabolites and genes. KEGG pathway enrichment of these genes showed 270 genes mapped to the biosynthesis of secondary metabolites and 192 genes mapped to the biosynthesis of antibiotics. This demonstrates the potent secondary metabolic capacity of strain YCSC6. Finally, a gene-metabolite correlation network was created based on the 575 continuously enhanced metabolites and 43 continuously up-regulated genes. This revealed 13 genes at the key position that mapped to a putative metabolic pathway associated with the biosynthesis of polyketides and caprylic acid, which contributes to the potent anti-parasitic activity of strain YCSC6. This comprehensive analysis of metabolomics and transcriptomics provides insights into the biosynthetic mechanisms of anti-parasitic compounds in strain YCSC6 and guides the exploitation of more anti-parasitic agents for aquaculture.
Keyword: metabolomics; transcriptomics; anti-parasitic; Salinivibrio proteolyticus; Uronema marinum
1 INTRODUCTION
The sustained and rapid growth of marine aquaculture globally has been accompanied by an increase in various parasites as disease agents. A dominant pathogenic ciliate in aquaculture,Uronemamarinum(Ciliophora, Scuticociliatida), is responsible for systemic scuticociliatosis in various marine f ish species, e.g., olive f lounder (Paralichthysolivaceus)(Jee et al., 2001), New Zealand groper (Polyprionoxygeneios) (Anderson et al., 2009), Atlantic salmon(Salmosalar) (Dyková et al., 2010), kelp grouper(Epinephelusbruneus) (Harikrishnan et al., 2011),black rockf ish (Sebastesschlegelii) (Whang et al.,2013), and turbot (Scophthalmusmaximus) (Du et al.,2019). Fish infected withU.marinumusually displayed skin lesions and loss of balance (Cheung et al., 1980;Anderson et al., 2009). Due to its high infectiousness, aU.marinumoutbreak can lead to signif icant mortalities and economic losses in aquaculture (Du et al., 2019).
Formalin, copper sulfate, hydrogen peroxide and other chemicals have traditionally been used in aquaculture for scuticociliatosis outbreaks(Harikrishnan et al., 2010; Du et al., 2019); however,their frequent use for parasitic control can be problematic, resulting in parasite resistance and deleterious eff ects, both environmental and in terms of human consumption. Extensive research has investigated the use of microorganisms and their secondary metabolites to control parasitic infections in aquaculture. Of particular importance are Natamycin, isolated fromStreptomycesgilvosporeuAXY-25, which kills allIchthyophthiriusmultif iliisat a concentration of 25.0 mg/L (Yao et al., 2019), and Nystatin, isolated fromStreptomycesgriseusSDX-4,which shows 100% eff ectivity againstI.multif iliistheronts and encysted tomonts at 6.0 mg/L (Yao et al.,2015). Therefore, to f ind high-eff ective and environment-friendly anti-parasitic agents, more than 1 000 strains of bacteria from the deep sea and off shore have been screened by anti-parasitic tests in vitro (Du et al., 2017). A bacterial strain, YCSC6, was screened out due to the potent anti-parasitic activity of its fermentation broth againstU.marinum. Strain YCSC6 was isolated from a saturated saltpan and identif ied asSalinivibrioproteolyticusin our previous study (Du et al., 2017, 2020). The fermentation broth of strain YCSC6 showed continuous enhancement of anti-parasitic activity throughout the incubation process within 72 h (Du et al., 2017), indicating that the active compounds in the fermentation broth were continuously accumulated.
Metabolomics, the identif ication and/or quantif ication of endogenous small molecules produced by a biological system at a specif ic time point, is an accepted and valuable tool in life sciences(Oldiges et al., 2007; Demarque et al., 2020).Substantial improvements of analytical hardware mean that metabolomics can be run routinely, and data have been used successfully to investigate genotype-phenotype relations of strains and mutants(Oldiges et al., 2007). Metabolomics is a valuable tool to analyze and identify the metabolites responsible for specif ic biological properties (Demarque et al.,2020). Additionally, due to the development of nextgeneration sequencing strategies, system biology and bioinformatics, the integration of metabolomics with transcriptomics is a novel approach that can investigate the metabolic mechanisms of organisms from a systems biology view with improved reliability.
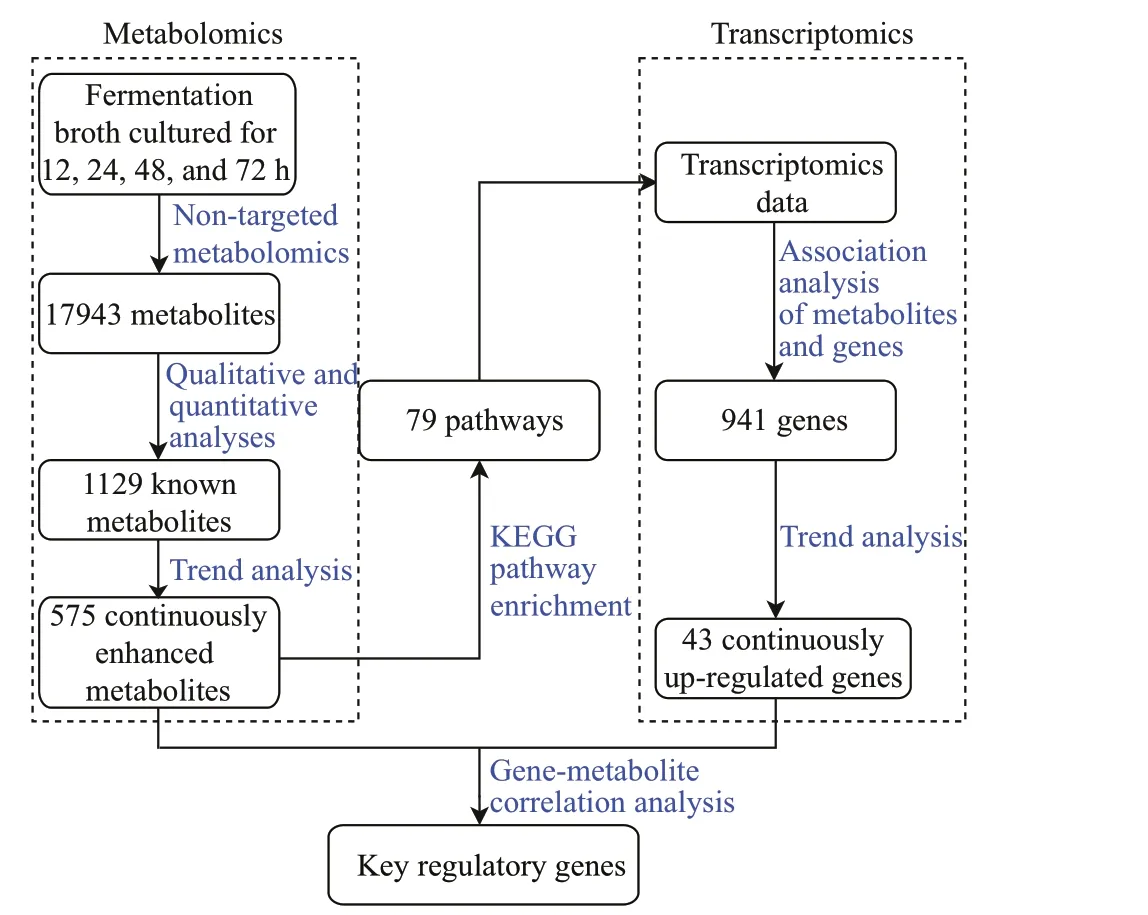
Fig.1 Overview of the metabolomic and transcriptomic analysis workf low in strain YCSC6
In this study, to reveal the biosynthetic mechanisms of anti-parasitic compounds in strain YCSC6, a combination of metabolomics and transcriptome over four diff erent time points (12, 24, 48, and 72 h) was performed. Then the continuously up-regulated genes and enhanced metabolites were screened out through the trend analysis. Based on these genes and metabolites, a gene-metabolite correlation network was created. Finally, a putative metabolic pathway associated with the biosynthesis of polyketides and caprylic acid was inferred. The integrated analysis of the metabolomics and transcriptome will lead to an in-depth knowledge of both the pool of metabolites and biosynthetic processes for the formation of antiparasitic compounds in strain YCSC6.
2 MATERIAL AND METHOD
The metabolomic and transcriptomic analysis workf low of strain YCSC6 is summarized in Fig.1.The metabolomics of strain YCSC6 was performed across four fermentation stages (12, 24, 48, and 72 h),detecting a total of 17 943 metabolites. Then 1 129 know metabolites were identif ied through the qualitative and quantitative analyses. A further trend analysis of the 1 129 known metabolites screened out 575 continuously enhanced metabolites, enriching in 79 metabolic pathways. In the same pathways, there were 941 genes being mapped through the association analysis of metabolites and genes. Then the trend analysis of the 941 genes screened out 43 continuously up-regulated genes associated with metabolism.Finally, a gene-metabolite correlation network was created based on the 575 continuously enhanced metabolites and 43 continuously up-regulated genes,presenting 13 key regulatory genes.
2.1 Sources of bacterial strain and parasite
Salinivibrioproteolyticusstrain YCSC6 was isolated from a saturated saltpan (120.82°E, 36.47°N)in China, and deposited in the China General Microbiological Culture Collection Center (CGMCC)as a patent strain, accession number 11730(information on patent strains is not released in CGMCC). The optimum growth conditions of strain YCSC6 are 37 °C, salinity of 30, and pH 6.0-7.0. The cultured liquid medium consisted of 10-g peptone(Oxoid, Hants, UK) and 5-g yeast extract (Oxoid,Hants, UK) per liter natural seawater (Du et al., 2020).
The parasite was isolated from the brain of an infected turbot and identif ied asU.marinumusing morphological characterization and molecular identif ication (Du et al., 2019). The cellular shape of this species is elongated, as an ellipsoid f lattened along the shorter axis, with a bluntly truncate anterior end and a lightly rounded posterior end. Cell size is(20-30) μm×(5-10) μm. The phylogenetic analysis based on 18S ribosomal RNA (18S rRNA) and cytochrome c cxidase subunit I (COX I) gene sequences showed this species was closest toU.marinumwith 100% conf idence (Du et al., 2019).For the axenic culture ofU.marinum, several ciliates were inoculated into 50-mL sterilized natural seawater containing 10 grains of rice, and maintained at 17 °C.The density ofU.marinumreached a maximum of 8 000 inds./L after 7 d.
2.2 Pre-experiment
Prior to preparing the samples for omics, the growth curve of strain YCSC6 was measured. Strain YCSC6 was inoculated in the liquid medium with 1%(v/v) of inoculum under aseptic conditions, followed by incubation at 37 °C with gentle shaking.Fermentation broth (2 mL) was removed every hour to measure the OD600. In addition, the in-vitro antiparasitic activity of the fermentation broth, againstU.marinumat diff erent fermentation stages, was measured as follows. Strain YCSC6 was inoculated in 2216E liquid medium, with 1% (v/v) of inoculum under aseptic conditions. This was followed by incubation at 30 °C with gentle shaking. The bacterial cells were removed from the fermentation broth after 12-72 h, at six-hour intervals, using 0.22-μm microf ilters (Millex-GP, Millipore, USA).Subsequently, the supernatant was co-cultured with ciliates at 17 °C for 0-12 h at hourly intervals. All the treatment groups and control groups were conducted in triplicate. The anti-parasitic activity was calculated using the following equation (Wang et al., 2010):Aa=(B-T)×100%/B, whereAais the anti-parasitic activity;Bis the mean number of surviving parasites in the control groups;Tis the mean number of the surviving parasites in the treatment groups.
2.3 Preparation of samples for transcriptomics and metabolomics
The pre-experiment study showed that the fermentation broth of strain YCSC6 demonstrated continuous enhancement of anti-parasitic activity within 72 h of incubation. Furthermore, the antiparasitic activity of the fermentation broth cultured for 12, 24, 48, and 72 h showed signif icant diff erences.Therefore, a comprehensive analysis of metabolomics and transcriptomics was performed, based on the fermentation broth cultured for these times. To prepare the samples for transcriptomics (groups Z12,Z24, Z48, and Z72), each group of fermentation broth was centrifuged at 4 000 r/min for 20 min. Bacterial cells (5 g) were collected and washed with phosphate buff er saline (PBS) three times then snap-frozen in liquid nitrogen. Three samples were used for replicates in each group. For the metabolomics (groups D12,D24, D48, and D72), the fermentation broth was f iltered using 0.22-μm microf ilters (Millex-GP,Millipore, USA) collecting 10 mL of supernatant,which were snap-frozen in liquid nitrogen. Nine samples were used for replicates in each group.
2.4 Transcriptome sequencing
Total RNA was extracted from strain YCSC6 using the TRIzol method (Life Technologies, CA, USA),according to the manufacturer’s protocol. RNA quality was verif ied as follows: (1) degradation and potential contamination were determined on 1%agarose gels; (2) purity (OD260/OD280 and OD260/OD230) was determined using a NanoPhotometer®spectrophotometer (IMPLEN, CA, USA); (3)integrity was measured using a Bioanalyzer 2100(Agilent, CA, USA). After the extraction of total RNA, ribosomal RNA (rRNA) was removed using the Ribo-ZeroTMRemoval Kit (Bacteria) (Illumina,CA, USA). The messenger RNA (mRNA) was then enriched and fragmented into short fragments using fragmentation buff er, and reverse transcribed into f irst-strand complementary DNA (cDNA) with random primers. The second-strand cDNA was synthesized by DNA polymerase I, RNase H, dNTP and buff er. The cDNA fragments were purif ied with the QiaQuick PCR extraction kit (Qiagen, Hilden,Germany), according to the manufacturer’s protocol,then end repaired with poly(A) added, and ligated to Illumina sequencing adapters. The ligation products were size-selected by agarose gel electrophoresis,PCR amplif ied and sequenced using an Illumina HiSeqTM2500 by Gene Denovo Biotechnology Co.(Guangzhou, China). The reads containing adapters and ploy-N, as well as the low-quality reads, were removed from the raw data to obtain clean reads. The clean reads were then aligned to the reference genome(GenBank accession number: CP039516-CP039517)using Bowtie2 (version 2.2.8) (Langmead and Salzberg, 2012) to identify known genes. Gene expression was calculated by RSEM (Li and Dewey,2011), and reads mapped to rRNA were removed. The gene expression level was further normalized using the fragments per kilobase of transcript per million mapped reads (FPKM) (Trapnell et al., 2010) method to eliminate the inf luence of gene lengths and amount of sequencing data on gene expression.
2.5 Metabolomic analysis
2.5.1 Metabolite extraction and LC-MS analysis
Filtered fermentation broth (500 μL) was placed in an Eppendorf tube and dried in a vacuum concentrator without heating. Subsequently, 300 μL of methanolwater (4꞉1 v/v) was added for re-dissolution, vortexed for 30 s, sonicated in an ultrasonic cooled bath for 3 min, centrifuged at 14 000 r/min for 10 min at 4 °C,and then 180 μL of supernatant was transferred into a fresh liquid chromatography-mass spectrometry (LCMS) glass vial for analysis. In the analysis process, a quality control (QC) sample was inserted into every ninth sample to monitor the repeatability of the analysis process.
LC-MS analysis was performed using an ultraperformance liquid chromatography (UPLC) system(Waters, Milford, USA) coupled to a 6520 seriesaccurate quadrupole time-of-f light mass spectrometer(Q-TOF/MS) system (Agilent, Santa Clara, CA,USA). Each sample (3 μL) was injected into an Acquity UPLC BEH C18column (100 mm×2.1 mm i.d., 1.7-μm particle size; Waters, Milford MA, US) for UPLC/MS analysis with a consistent column temperature of 50 °C. The mobile phases consisted of deionized water with 0.1% formic acid for canal A and acetonitrile with 0.1% formic acid for canal B. The f low rate was set at 0.4 mL/min. A linear gradient for elution was set as follows: 1%-1% B for 0-1.0 min;1%-20% B for 1.0-5.5 min; 20%-30% B for 5.5-6.0 min; 30%-35% B for 6.0-8.5 min; 35%-70% B for 8.5-10.5 min; 70%-100% B for 10.5-11.0 min;100%-100% B for 11.0-13.0 min; 100%-1% B for 13.0-13.1 min; and 1%-1% B for 13.1-15.0 min.
The acquisition of a mass spectrum signal was adopted in the positive-ion (ESI+) and negative-ion(ESI-) modes. The electrospray capillary voltage,injection voltage and impact voltage were 1.0 kV,40 V, and 6 eV, respectively. The temperature of the ion source and dissolvent were 120 °C and 500 °C,respectively. The carrier gas f low was 900 L/h, and the scanning range of mass spectrometry was 50-1 000m/z. The scanning time and interval time were 0.1 and 0.02 s, respectively.2.5.2 Qualitative and quantitative analyses of metabolites
The base peak chromatogram (BPC) of all samples was checked visually, and then the mass spectrometry raw data were processed by progenesis QI (Waters,Milford, USA) for baseline f iltering, peak recognition,integration, retention time correction, peak alignment,and normalization. Finally, a data matrix, including retention time, mass charge ratio and peak intensity was obtained. The obtained peaks were identif ied using progenesis QI (Waters, Milford, USA) with the public metabolite databases: the Human Metabolome Database (HMDB) (Wishart et al., 2013) and LIPID MAPS Structure Database (Sud et al., 2007).
2.6 Trend analysis of metabolites
Short Time-series Expression Miner (STEM)software (Ernst and Bar-Joseph, 2006) was used to display the trends across four stages (D12, D24, D48,and D72) and analyze the expression trend of all metabolites, based on the quantitative value of integration in each sample. All metabolites were divided into 20 modules according to the expression pattern, and thePvalue of each module was calculated using the Permutation Test method (Oden and Wedel,1975).
2.7 Association analysis of metabolites and genes
Metabolites and genes in the same pathways always dysregulated together, so a pathway-based approach was used to integrate diff erent levels of omics in the biological process. After the trend analysis, the metabolites with continuous upregulation trend were selected to map to the Kyoto Encyclopedia of Genes and Genomes (KEGG)database (http://www.genome.jp/kegg/pathway.html)for pathway enrichment analysis. The genes enriched in the same pathways were then selected for Gene Ontology (GO) enrichment analysis using the OmicShare tools (http://www.omicshare.com/tools)and trend analysis using STEM software (Ernst and Bar-Joseph, 2006).
To further screen out the genes at the key association position, a Pearson correlation coeffi cient model was constructed, based on the gene expression and metabolite abundance (Bartel et al., 2015). Gene and metabolite pairs were ranked in descending order of absolute correlation coeffi cients, and the top 250 pairs of genes and metabolites (absolute Pearson correlation>0.5) were used for the gene-metabolite correlation network analysis using igraph packages in R project(Csárdi and Nepusz, 2006).
2.8 In-vitro anti-parasitic activity assay of the candidate compounds against U. marinum
The candidate compounds to be tested include erythromycin, piceatannol, and caprylic acid. Each compound was dissolved in dimethyl sulfoxide(DMSO) to form stock solutions. Then, 100 individuals ofU.marinumwere dispensed into a 24-well tissue culture plate, the f inal test concentration of each compound was set according to the rangef inding test. Negative controls were set containing the same amount of DMSO as the maximum concentration test group, and the highest concentration of DMSO in the treatment was less than 3% (preliminary analysis with 3% DMSO showed no activity againstU.marinum). The plates were incubated at 17 °C for 24 h and the parasites were considered dead when the specimen did not show any motility with a light touch. All the test and control groups were conducted in triplicate. The median eff ective concentration (EC50) of each compound was calculated at the 95% conf idence interval by Probit analysis (Finney, 1971).
2.9 RT-qPCR verif ication of transcriptome data
To verify the transcriptome data, the expression levels of 13 candidate genes under four culture phases(12, 24, 48, and 72 h) were analyzed by real-time quantitatire reverse transcription-PCR (RT-qPCR).TherecA gene was selected as the internal reference gene (Dourado et al., 2013). All the gene-specif ic primers were designed using Primer 5.0 (Zhai et al.,2008) (Supplementary Table S1). The reaction had a f inal volume of 20 μL, containing 10 μL of 2× SYBR Premix Ex Taq II (TaKaRa, Dalian, China), 0.4 μL of 50× ROX reference dye II (TaKaRa, Dalian, China),0.8 μL each of forward and reverse primers, 2-μL cDNA, and 6-μL ddH2O. The quantitative reaction was performed on an ABI 7500 RT-PCR System using the two-step method, under the following conditions: 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s and 55 °C for 34 s, and f inally 95 °C for 15 s, 60 °C for 30 s and 95 °C for 15 s. The specif icity of amplif ication was assessed by melt curve analysis.The relative gene expression levels were calculated using the 2-ΔΔCtmethod (Livak and Schmittgen, 2001).All RT-qPCR experiments were performed with three biological and three technical replications.
3 RESULT
3.1 Pre-experiment and sample preparation
The growth curve (Fig.2a) shows that strain YCSC6 reached the stationary-phase after 12-h incubation. The anti-parasitic assay (Fig.2b) shows the anti-parasitic activity of the fermentation broth enhanced continuously over 72 h. Furthermore, the anti-parasitic activity of the fermentation broth cultured for 12, 24, 48, and 72 h exhibited signif icant diff erences. Based on these results, the fermentation broths cultured for 12, 24, 48, and 72 h were selected for performing the comprehensive analysis of metabolomics and transcriptomics.
3.2 Metabolomics analysis
A two-dimensional (2D) principal component analysis (PCA) score plot of all samples revealed no outliers in this study (Fig.3), and the tightly clustered QC samples conf irm the detection stability. A total of 17 943 LC-MS spectral peaks were detected in all samples, with 1 129 metabolites successfully matched with compounds in the public databases, based onm/zand normalized migration time. All the 1 129 known metabolites are listed in Supplementary Table S2.There is mainly a diverse range of fatty acyls (253 metabolites), polyketides (161 metabolites),glycerophospholipids (147 metabolites), sterol lipids(95 metabolites), prenol lipids (69 metabolites), and amino acid derivatives (66 metabolites).

Fig.2 The results of the pre-experiment study
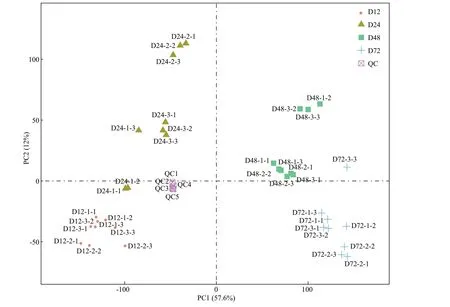
Fig.3 The 2D PCA score plot of the metabolite samples of strain YCSC6 and quality control (QC)
3.3 Trend analysis of the metabolites
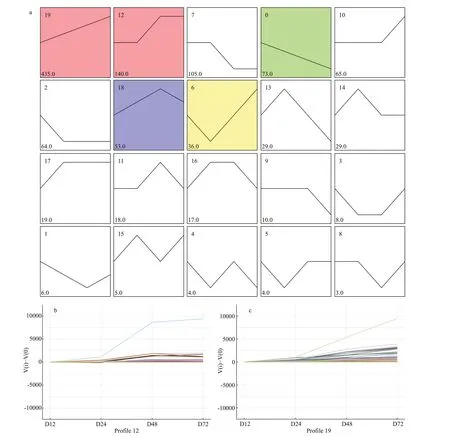
Fig.4 Trend analysis of the metabolites across four fermentation stages (12, 24, 48, and 72 h) of strain YCSC6
As shown in Fig.4a, all 1 129 known metabolites clustered into 20 prof iles based on their expression modulation. Of these, 737 metabolites clustered into f ive signif icant prof iles (P<0.05, colored squares),including two up-regulated patterns (prof ile 19 and prof ile 12), one down-regulated pattern (prof ile 0),one up-down-regulated pattern (prof ile 18) and one down-up-regulated pattern (prof ile 6). According the anti-parasitic activity phenotype of the fermentation broth (continuous enhancement over 72 h), the metabolites clustered into prof ile 19 (435 metabolites)(Fig.4c) and prof ile 12 (140 metabolites) (Fig.4b)were selected for further analysis. Among the 575 upregulated metabolites (Supplementary Table S3),there were mainly 153 fatty acyls, 69 polyketides, 41 prenol lipids, 33 sterol lipids, 27 amino acids and derivatives, as well as three f lavonoids. In addition,the KEGG pathway enrichment analysis of these metabolites showed a total of 125 metabolites mapped to 79 pathways as follows: metabolic pathways (96 metabolites), biosynthesis of secondary metabolites(49 metabolites), glycerophospholipid metabolism(22 metabolites), microbial metabolism in diverse environments (21 metabolites), biosynthesis of antibiotics (16 metabolites), biosynthesis of amino acids (14 metabolites), 2-oxocarboxylic acidmetabolism (12 metabolites), arachidonic acid metabolism (10 metabolites), ABC transporters (10 metabolites), phenylalanine metabolism (9 metabolites), tryptophan metabolism (8 metabolites),and tyrosine metabolism (6 metabolites)(Supplementary Table S4).
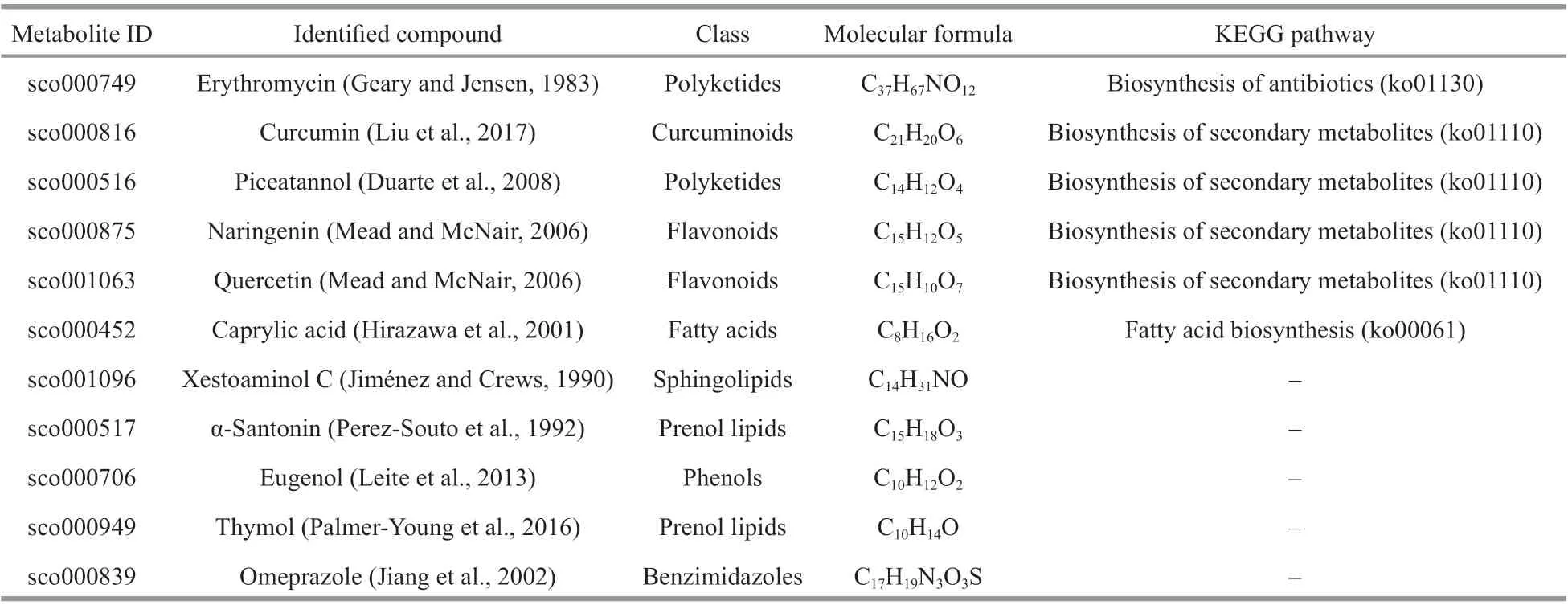
Table 1 Metabolites with anti-parasitic activity
According to the literature, 11 metabolites(compounds) reported to have anti-parasitic activity were manually screened out from the 575 up-regulated metabolites. Details of the 11 compounds are listed in Table 1. In addition, erythromycin, piceatannol, and caprylic acid were subjected to an anti-parasitic activity assay againstU.marinum. The resulting EC50(24 h) values of erythromycin and piceatannol were 31.64 mg/L and 4.79 mg/L, respectively. Caprylic acid showed 100% activity againstU.marinum, with a volume ratio of 1꞉2 000 after 24-h incubation.
3.4 Association analysis of metabolites and genes
After performing metabolite pathway enrichment analyses on 79 pathways, a total of 941 genes(Supplementary Table S5) mapped to the same pathways, were analyzed further. The KEGG pathway enrichment analysis mapped 552 of these genes to metabolic pathways: 270 genes mapped to secondary metabolites biosynthesis, 192 genes mapped to antibiotic biosynthesis, 156 genes mapped to microbial metabolism in diverse environments, and 139 genes mapped to the two-component system.
Further, GO enrichment of the 941 genes showed there were 742, 714, and 245 genes enriched to molecular function, biological process, and cellular component, respectively (Fig.5). The directed acyclic graph (DAG) of molecular function showed the highest degree of enrichment was in catalytic activity(608 genes) (Fig.6). Further enrichment analysis of these genes showed they were mainly involved in transferase activity (195 genes), oxidoreductase activity (148 genes), lyase activity (65 genes), and ligase activity (51 genes). The trend analysis of these genes manually screened out 43 continuously upregulated genes (Supplementary Table S6) associated with metabolism.
The gene-metabolite correlation network (Fig.7),based on the 575 continuously up-regulated metabolites and 43 continuously up-regulated genes associated with metabolism showed 13 genes were at the key association positions, which might play an important role in the metabolic regulation of strain YCSC6. Details of these genes are listed in Table 2.Among the 13 genes, f ive genes were related to secondary metabolites biosynthesis (ko01110) and four genes were related to antibiotic biosynthesis(ko01130).
3.5 RT-qPCR verif ication of transcriptome data
The gene expression at 12 h was set as one, and the relative gene expression was calculated for 24, 48,and 72 h. As shown in Fig.8, the relative expression of all nine candidate genes presented a continuously upregulated trend across these four stages (D12, D24,D48, and D72), which was similar to the transcriptomic results.
4 DISCUSSION

Fig.5 Gene ontology (GO) enrichment analysis of 941 target genes

Table 2 Details of 13 key genes
The global rise of f ish aquaculture has been accompanied by an increase in parasites as causative disease agents. Extensive research has been conducted on the utilization of microorganisms, and their secondary metabolites, to control parasitic infections in aquaculture (Yao et al., 2015, 2019). In our previous study, the fermentation broth ofS.proteolyticusstrain YCSC6 was shown to have potent anti-parasitic activity againstU.marinum, which improved continuously over 72 h. In this study, to identify the biosynthetic mechanism of anti-parasitic activity of strain YCSC6, a comprehensive analysis of metabolomics and transcriptomics was performed at four diff erent time points.
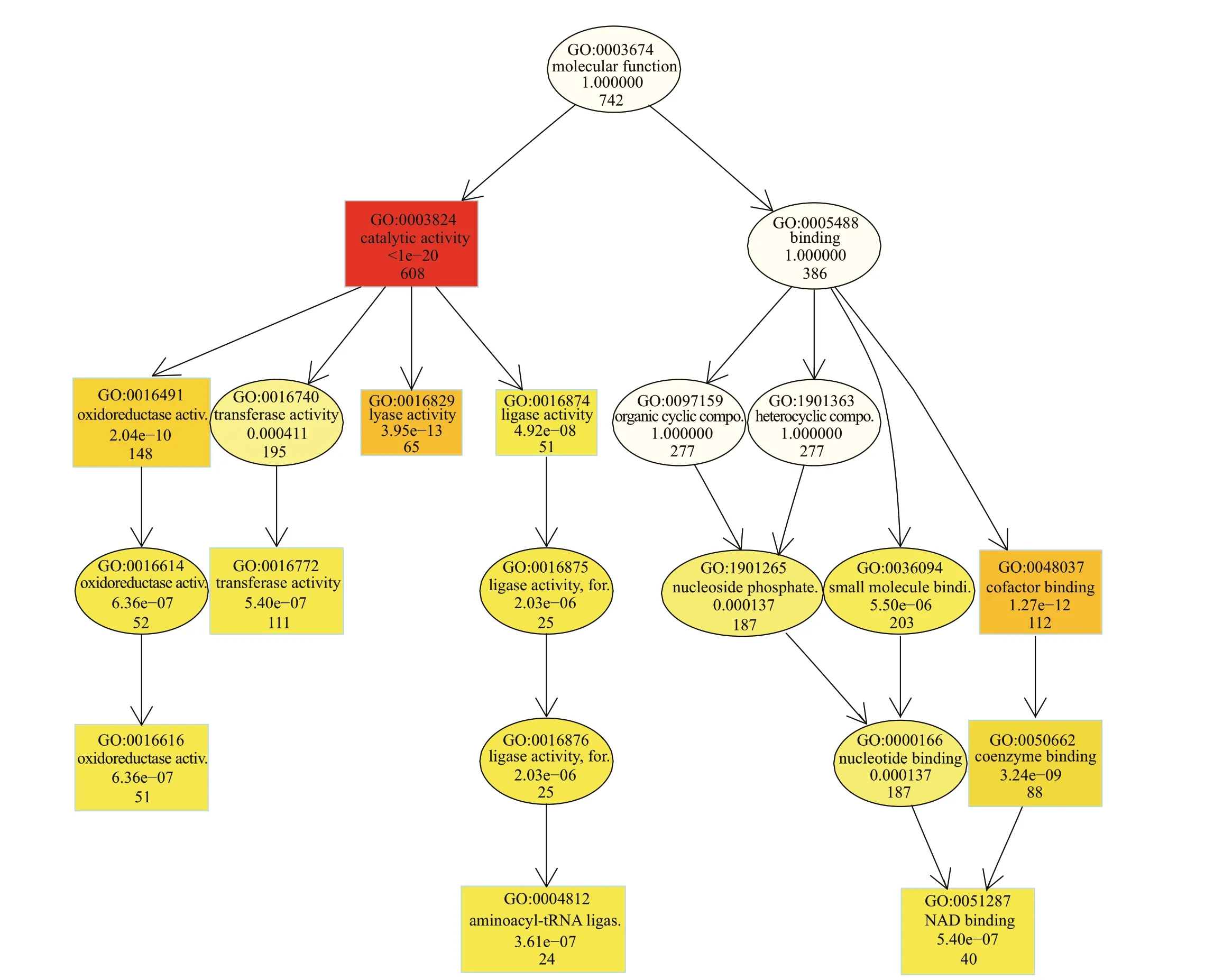
Fig.6 Directed acyclic graph (DAG) of GO enrichment of genes clustered in molecular function
Non-targeted metabolomics detected 17 943 metabolites, including 1 129 known metabolites(Supplementary Table S2). A further trend analysis of these known metabolites screened out 575 continuously up-regulated metabolites (Supplementary Table S3),including 69 polyketides (Bender et al., 1999; Kossuga et al., 2008) and three f lavonoids (Mead and McNair,2006), which could be responsible for the potent antiparasitic activity of strain YCSC6. Previous studies have found 11 metabolites (Table 1) that have antiparasitic activity, and which might contribute to the activity of strain YCSC6 againstU.marinum. In addition, the KEGG pathway enrichment analysis of the 575 continuously up-regulated metabolites showed a total of 125 metabolites mapped to 79 pathways(Supplementary Table S4). Among these metabolites,49 metabolites mapped secondary metabolites biosynthesis and 16 metabolites mapped to antibiotic synthesis. This indicates the potential secondary metabolic capacity of strain YCSC6 for the production of anti-parasitic active compounds.
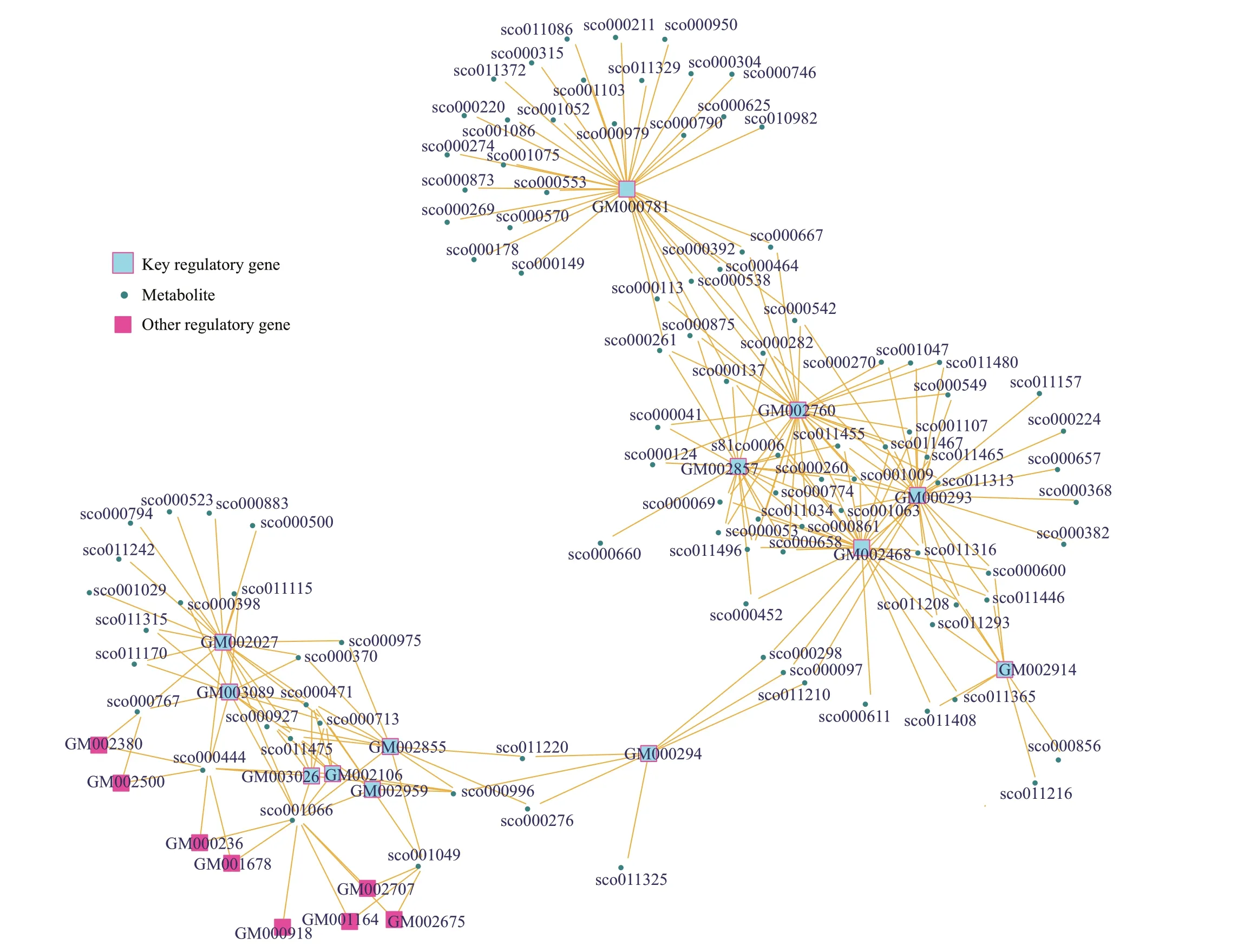
Fig.7 The gene-metabolite correlation network of metabolites and genes
Metabolites and genes involved in the same biological process always regulated together: this provides a method for performing an associate analysis to reveal the regulatory mechanisms between them (Yang et al., 2017). Using this methodology, a total of 941 genes were mapped to the 79 pathways(Supplementary Table S5) with 270 genes mapping to secondary metabolite biosynthesis and 192 genes mapping to antibiotic biosynthesis, which indicated the potential secondary metabolic capacity of strain YCSC6. The GO enrichment analysis of the 941 genes also showed that 608 genes were associated with catalytic activity, with the highest degree of enrichment (Fig.6): the abundant catalytic enzymes ensured the secondary metabolic capacity of strain YCSC6. Furthermore, 13 key genes (Table 2) were screened out through the gene-metabolite correlation analysis (Fig.7). These genes might play an important role in the regulation of the anti-parasitic activity of strain YCSC6. Among the 13 key genes, genes GM000293 (ilvI) and GM000294 (ilvH) encoded acetolactate synthase, which is a key enzyme of branched-chain amino acids (valine, leucine and isoleucine) biosynthesis in bacterial cells (Sun et al.,2008). Valine, leucine, and isoleucine are precursors for polyketide secondary metabolite synthesis(Craster et al., 1999): the erythromycin biosynthesis precursors, propanoyl-CoA and methylmalonyl-CoA,are derived from the branched-chain amino acids(Hopwood and Sherman, 1990; Marsden et al., 1994).In this study, erythromycin (macrolides and lactone polyketides) and piceatannol (aromatic polyketides)were detected in the fermentation broth of strain YCSC6, and they showed anti-parasitic activity againstU.marinum. According to the associate metabolic pathways analysis of metabolites and genes of strain YCSC6, it can be inferred that strain YCSC6 synthesized L-valine and L-isoleucine from 2-oxobutanoate and pyruvate under the catalysis of acetolactate synthase and other enzymes(Supplementary Fig.S1). L-valine and L-isoleucine were then degraded through the valine, leucine, and isoleucine degradation pathway (Supplementary Fig.S2), which provided precursors for the biosynthesis of 12-, 14- and 16-membered macrolides and biosynthesis of type II polyketide backbone. Finally,erythromycin, piceatannol and other polyketides were synthesized by strain YCSC6: these compounds contributed to the potent anti-parasitic activity of strain YCSC6, the EC50(24 h) values of erythromycin and piceatannol were 31.64 and 4.79 mg/L,respectively. The ectoine hydrolase, encoded by gene GM002914 (doeA), is involved in glycine, serine, and threonine metabolism (Supplementary Fig.S3), which produced 2-oxobutanoate for the biosynthesis of L-isoleucine. Genes GM002106 (gshA) and GM002468 (met17) are involved in cysteine and methionine metabolism (Supplementary Fig.S4),which produced 2-oxobutanoate and pyruvate for the biosynthesis of L-isoleucine and L-valine. Genes GM002855 (gnd), GM002857 (zwf) and GM003026(edd) encode the key enzymes of the pentose phosphate pathway (Supplementary Fig.S5), which produce pyruvate for the biosynthesis of L-isoleucine and L-valine, as well as 5-phospho-α-D-ribose 1-diphosphate (PRPP) for purine metabolism(Supplementary Fig.S6), pyrimidine metabolism(Supplementary Fig.S7) and histidine metabolism(Supplementary Fig.S8). In addition, genes GM002760 (nrdD) and GM002959 (udp) are involved in pyrimidine metabolism (Supplementary Fig.S7),which produce methylmalonate and (R)-3-aminoisobutyrate for valine, leucine and isoleucine degradation. All of the above pathways together enhance the biosynthesis of metabolite precursors for the biosynthesis of polyketides.
Gene GM002027 (fabZ) mapped to the pathway of fatty acid biosynthesis (Supplementary Fig.S9),encoding the 3-hydroxyacyl-[acyl-carrier-protein]dehydratase that regulates the biosynthesis of caprylic acid. Meanwhile, pyruvate metabolism (Supplementary Fig.S10) provided the precursors for caprylic acid biosynthesis (malonyl-CoA). Metabolomics identif ied metabolite sco000452 of strain YCSC6 as caprylic acid: this medium-chain fatty acid had been shown to have 100% activity againstU.marinum, with a volume ratio of 1꞉2 000 after 24-h incubation. It can, therefore,be inferred that the continuous enrichment of caprylic acid contributes to the potent anti-parasitic activity of strain YCSC6.
From the metabolic pathways analysis, a putative comprehensive metabolic pathway associated with the active compound biosynthesis was inferred(Fig.9). This indicates that, under the control of key genes, strain YCSC6 produces abundant polyketides and caprylic acid, giving the fermentation broth its potent anti-parasitic activity.
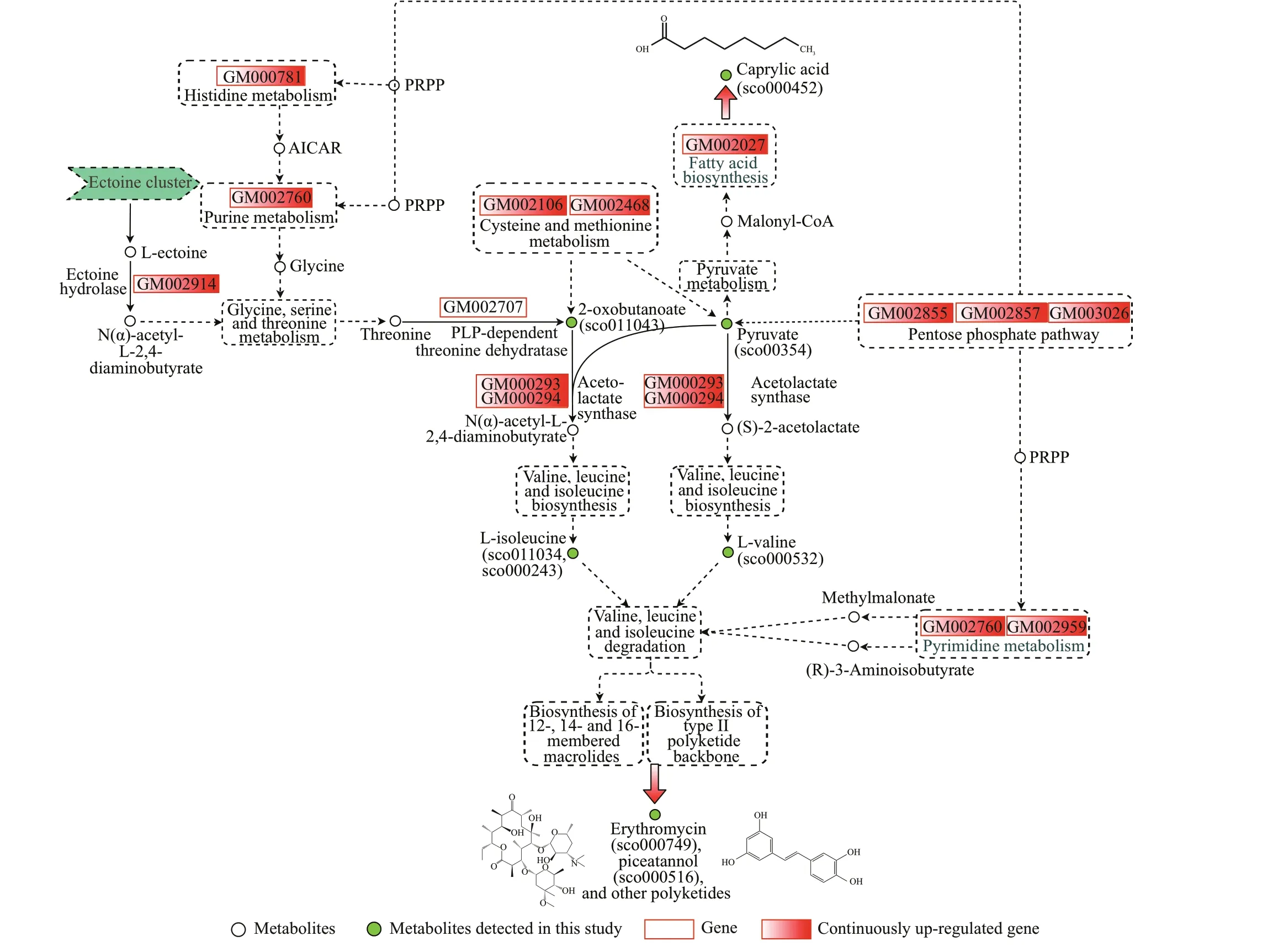
Fig.9 Putative metabolic pathways for the biosynthetic of active compounds (polyketides and caprylic acid) in strain YCSC6
5 CONCLUSION
The present study investigated the biosynthetic mechanism of anti-parasitic activity ofSalinivibrioproteolyticusstrain YCSC6 through a comprehensive analysis of metabolomics and transcriptomics. The results indicate that strain YCSC6 produces abundant polyketides and caprylic acid through a putative comprehensive metabolic pathway under the regulation of 13 key genes. This provides insights into the molecular mechanisms underlying the potent anti-parasitic activity of strain YCSC6 and guides the exploitation of more anti-parasitic agents for aquaculture.
6 DATA AVAILABILITY STATEMENT
The raw reads of the transcriptome sequencing have been deposited in NCBI Sequence Read Archive(SRA) under the accession numbers SRR13084579-SRR13084590.
 Journal of Oceanology and Limnology2022年1期
Journal of Oceanology and Limnology2022年1期
- Journal of Oceanology and Limnology的其它文章
- The adjoint-based Two Oceans One Sea State Estimate(TOOSSE)*
- Structure and formation of the South Yellow Sea water mass in the spring of 2007*
- Lagrangian eddies in the Northwestern Pacif ic Ocean*
- Seasonal variability in dissolved oxygen in the Bohai Sea,China*
- In-situ experiments reveal mineralization details of porphyry copper deposits
- Chemical composition and Pb(II) binding of dissolved organic matter in a hypersaline lake in China*