
Transcriptional response of Microcystis aeruginosa to the recruitment promoting-benthic bacteria *
2022-03-05 09:35JuanWANGYuandePENGZhiWANGWanshengZOUXianjinPENGQishengSONG
Juan WANG , Yuande PENG , Zhi WANG , Wansheng ZOU , Xianjin PENG ,Qisheng SONG
1 College of Life Sciences, Hunan Normal University, Changsha 410081, China
2 Institute of Bast Fiber Crops, Chinese Academy of Agricultural Sciences, Changsha 410205, China
3 Department of Life Science, Hunan University of Arts and Science, Changde 415000, China
4 Division of Plant Sciences, University of Missouri, Columbia 65211, MO, USA
Abstract Blooms of Microcystis aeruginosa occur frequently in many freshwater ecosystems around the world, but the mechanism of recovery has not been fully understood. In our previous study, three benthic bacterial species (E.sp013, Ba.spD06, and Ba.spD24) were identif ied capable of promoting the recruitment of M. aeruginosa. Here, we further investigated the transcriptional response of M. aeruginosa to the benthic bacteria in early phase of recruitment by means of RNA-Seq analysis. In total, 5 803 803 unigenes on average length of 404 bp were obtained from the transcriptome of M. aeruginosa. There were 54 982 unigenes identif ied as benthic bacteria-responsive unigenes based on the expression level analysis. Results of the protein-protein interaction analysis (PPI) show that the hub genes of the benthic bacteria responsive unigenes mediated network were ribosomal proteins of 30S and 50S, and the most signif icant functional module of the network was related to the ribosome. Both the unigenes encoding the translation initiation factors (IF-2, IF-3) and elongation factors (lepA, fusA, and tufA) were up-regulated to respond benthic bacteria. Therefore, it indicates that the benthic bacteria have a positive inf luence on activating the ribosome during the early recovery stage of M. aeruginosa.
Keyword: Microcystis aeruginosa; ribosome; benthic bacteria; chlorophyll a
1 INTRODUCTION
Microcystisis one of the most notorious phytoplankton species, acting as a dominant species on the lake eutrophication (Mohamed et al., 2003;Robson and Hamilton, 2004; Gan et al., 2012).Studies suggested thatMicrocystiscould be the dominant species in the aquatic ecosystem, mainly due to its ecological advantages over other phytoplankton species (Paerl et al., 1985; Kruk et al., 2017; Esterhuizen-Londt et al, 2018). For example,Microcystisis characterized with sheaths and mucilage, which increases the f loating velocity and resistance of cells (Cyr and Curtis, 1999; Kearns and Hunter, 2000; Wu and Kong, 2009; Yamamoto et al., 2011). In eutrophication water,Microcystiswas able to f loat to water surface to capture light and CO2, and outcompete the growth of other planktonic algae (Dokulil et al., 2000; Pfeifer, 2012). In addition,Microcystiscould actively enrich phosphorus in nutrient-limited water by forming polyphosphate granules. They can be reproduced using such stored phosphorus under favorable conditions for growth (Jacobson and Halmann,1982). Signif icantly,Microcystisoverwinters in benthic sediments in the form of hypopus,outcompete in survival rate of other planktonic algae, and provide the inoculum for the next bloom(Verspagen et al., 2004, Misson et al., 2012). Several investigations have suggested that the germination of hypopus had provided the f irst propagules for development of newMicrocystispopulations(Preston et al., 1980, Head et al., 1998). Hence,recruitment from resting stages might be an important process ofMicrocystispopulation dynamics.
It is of particular importance to understand the recruitment of rest stages for the prediction and mitigation ofMicrocystisblooms. Recently, multiple abiotic and biotic factors have been recognized able to aff ect the recruitment ofMicrocystis. The abiotic factors, including altered temperature, light conditions, and concentrations of inorganic nutrients were contributed to the recruitment ofMicrocystis(Ståhl-Delbanco et al., 2003; Karlsson-Elfgren et al.,2004). Reynolds and Jaworski (1978) suggested thatMicrocystiswould be active in sediments when temperature of deep lakes is 7-8 °C, and would grow rapidly at 15 °C. Brunberg and Blomqvist (2003)indicated that enhanced light and temperature were important for triggering the growth of benthicMicrocystiscolonies, and the eff ects of biotic factors such as benthic fauna on the recruitment ofMicrocystisresting stages were also reported. The benthic invertebrates were found to stimulate the recruitment of freshwater cyanobacteria, by probably increasing nutrient availability in the pore water (Ståhl-Delbanco and Hansson, 2002). Sediment is a complex microenvironment that contains abundant benthic bacteria (Risgaard-Petersen et al., 2015; Zhou et al.,2017). Xing et al. (2011), Li et al. (2012), and Shao et al. (2014) reported signif icant association between bacterial communities andMicrocystisblooms. Wu et al. (2019) found that the diversity and composition of the benthic microbial communities were changed after the addition ofMicrocystisbiomass. In an earlier study of ours, three dominant strains of bacteria(named E.sp013, Ba.spD06, and Ba.spD24) were isolated from sediment of Chongtian Lake and identif ied able to accelerate the recovery ofMicrocystisaeruginosa(Zou et al., 2018). However, less is known on the molecular response ofMicrocystisto those recruitment-promoting factors. In this study, we investigated the transcriptional response ofM.aeruginosatriggered by benthic bacteria using the RNA-Seq analysis, and underlined the role of benthic bacteria in the recruitment process ofM.aeruginosain the early phase.
2 MATERIAL AND METHOD
2.1 Preparation of recruitment experiments
The Chongtian Lake (Changde City, Hunan Province, China) is often suff ered from blooms caused byM.aeruginosain the past few years (Zou et al.,2017). As described in Zou et al. (2018), three dominant benthic bacteria (E.sp013, Ba.spD06, and Ba.spD24) that separated from the sediment of Chongtian Lake were found able to induce the recruitment ofM.aeruginosa. In this study, the simulation experiments on the recruitment ofM.aeruginosacells were performed in the presence or absence of the dominant benthic bacteria. As described in our previous study (Zou et al., 2018), the sediment,lake water, andM.aeruginosacell were sampled from the lake. The sediment was sampled using the KC Kajak sediment corer (6.5-cm diameter, KC,Denmark), and screened by a f ilter (sieve size 125 μm).The lake water was modif ied by adding NaNO3(0.16 g/L) and K2HPO4(0.07 g/L) to provide enough nutrition for the growth of bacteria andM.aeruginosa.TheM.aeruginosacell was expanding incubated in BG11 liquid media (provided by the Chinese Academy of Sciences, 25 °C, 50 μmol photons/(m2·s)), then placing at low temperature and dark for 3.5 months,and f inally obtained the hypopus ofM.aeruginosa.Three dominant benthic bacteria were cultured by the beef peptone liquid medium and mixed with the volume ratio of 1꞉1꞉1 (f inal volume: 10 mL), then added withM.aeruginosacell solution in the same volume (10 mL). 10 mL of lake water instead of mixed bacteria was added as the control. Both the whole mixed solution and sediment were transferred into sterilized cylindrical glassware (SCG: diameter 15 cm,height 50 cm) and diluted to 7 L with sterilized lake water. All SCGs were placed in the artif icial chamber(MMM Climacell, Germany) at the irradiance of 15 μmol photons/(m2·s), the temperature of 15 °C, and illumination time 12 h꞉12 h (light꞉dark). TheM.aeruginosarecruit for 3 days (control: CK3;treated: TR3), 5 days (control: CK5, treated: TR5),and 7 days (control: CK7; treated: TR7) were collected by centrifugation (2 000 r/min, 5 min) and used for RNA-Seq analysis.
2.2 Determination of chlorophyll a
TheM.aeruginosawere collected from the sediment and overlying water with the diff erent recruited days respectively, according to the method of Verspagen et al. (2004). The chlorophyll-aconcentration ofM.aeruginosawas determined according to the method of Berrendero et al. (2016).All data were presented as mean±SD, and conducted for one-way ANOVA by SPSS 17.0 software.
2.3 RNA-Seq analysis
2.3.1 Sequencing and annotation
RNA extraction and library construction were conducted using the TRzolRReagent (Thermo Fisher Scientif ic, USA) and NEBNext®Ultra™ RNA Library Prep Kit for Illumina®(NEB, USA),respectively. All samples were sequenced on the Illumina sequencing platform (HiSeqTM 2500), and generated paired-end reads with a length of 150 bp/125 bp. Raw reads were assembled to transcript sequences by Trinity (Grabherr et al., 2011). The public databases were applied for functional annotation of unigenes, including Nr, Swiss-Prot,KEGG, and GO.
2.3.2 Gene expression analysis
All of the clean reads were mapped back onto the assembled transcriptome reference sequences through RSEM, after which the read count value of each gene was calculated (Li and Dewey, 2011). Then the FPKM algorithm was used to normalize the read count value of each gene (Trapnell et al., 2010). Finally,diff erentially expressed genes were screened by software DESeq based on the negative binomial distribution. A threshold for adjustP-value (q)<0.05 and absolute value of Log2 fold change value >1 was used to determine signif icant diff erences in gene expression. Moreover, the KEGG enrichment analysis was performed to describe the function of diff erentially expressed genes (DEGs) with thresholdq<0.05.
2.3.3 Protein to protein interaction analysis (PPI)
The associations between the proteins and the proteins encoded by DEGs were determined using the STRING database (http://string-db.org/), with setting the string score at 700 (Seebacher and Gavin,2011). Cytoscape software was applied for visualization and analysis of the network. The Cytohubba plugin of Cytoscape was applied to identify the hub genes in the network, based on the MCC algorithm. MOCDE plugin of Cytoscape was applied to screen modules from the PPI network with degree cutoff =2, node score cutoff =0.2, k-core=2,and max. Depth=100 (Talebi et al., 2018) Also, the functional modules were chosen with the cutoffvalues of intra connection nodes≥6 and node score≥4.0.
2.4 RT-qPCR analysis
A total of 5 diff erentially expressed transcription factors (TFs) were selected and quantif ied by realtime RT-PCR (RT-qPCR). The First Strand cDNA Synthesis Kit was used for inverse transcription according to the manufacturer’s instructions(TINGEN, China). Real-time PCR was performed using the SYBR Premix EX Taq (TINGEN, China) by the ABI 7500 Real-time PCR System (Thermo Fisher Scientif ic, USA). In this study, the expression levels of the target unigene were normalized by 16S ribosomal RNA and quantif ied using the 2-ΔΔCtmethod(Livak and Schmittgen, 2001).
3 RESULT
3.1 Overview of transcriptome from the M. aeruginosa
As described in our previous study (Zou et al.,2018), the concentration of chlorophylla(Chla) in the sediment was decreased with time, while those in the overlying water was increasing, which suggested thatM.aeruginosaf loated from sediment into the overlying water and proliferating rapidly there. The recovery effi ciency ofM.aeruginosain the benthic bacteria-treated group was higher than that in control.
RNA-Seq analysis was conducted forM.aeruginosaat the early phase of recruitment(3-7 days). We obtained 5 803 803 unigenes with an average length of 404 bp. The N50 value of unigenes was 428 bp. The unigenes in length of 200 to 500-bp sequences represented the highest proportion,followed by 500 to 1 000-bp sequences (Fig.1).
3.2 Gene expression and function analysis
Diff erential expression analysis was conducted between the samples from the diff erent recovery days.For control ones, there were 1 and 76 843 unigenes up-regulated in the comparisons CK5 vs. CK3, and CK7 vs. CK5 respectively. For benthic bacteria treated ones, there were 71 570 and 104 351 unigenes up-regulated in the comparisons TR5 vs. TR3, and TR7 vs. TR5 respectively (q<0.05, |log2.Fold_change|>1), showing the more active status in expression of genes. Results from the RT-qPCR showed that the expression of selected unigenes was consistent with transcriptome analysis (Supplementary Table S1).
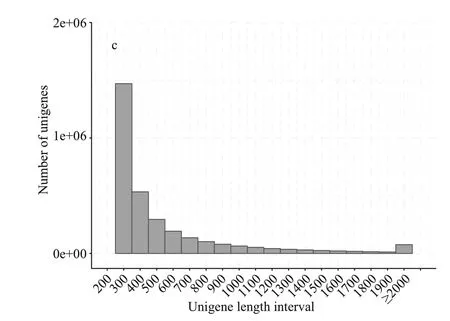
Fig.1 Overview of transcriptome from the Microcystis aeruginosa
Interestingly, a set of unigenes (54 982 unigenes)was up-regulated in the comparison CK7 vs. CK5 and was also up-regulated in comparison TR5 vs. TR3,which suggested that these unigenes would express earlier inMaeruginosaunder the inf luence of benthic bacteria (named benthic bacteria responsive-unigenes,marked with grayness, Fig.2a). KEGG enrichment analysis was conducted for these benthic bacteria responsive unigenes, found the “Ribosome” was the most enriched pathway and followed by “Cell cycle -Caulobacter” and “Oxidative phosphorylation”. This suggested that functions of these unigenes were involved in the ribosome, mitosis, and energy metabolism ofM.aeruginosacells (Fig.2b).
3.3 Protein to protein interaction analysis (PPI) for benthic bacteria responsive unigenes
The hub genes and core functions of benthic bacteria-responsive unigenes were characterized based on the PPI analysis. The PPI network was consisted of 1 092 interactions and 191 proteins,providing the overview of functional links between proteins encoded by those responsive unigenes(Fig.3a). From the total PPI network, transcription factor sigma A (sigA, MAE_54470, marked with red),translation initiation factors IF-2 (MAE_14330) and IF-3 (MAE_30180), elongation factors lepA(MAE_39840), fusA (MAE_42760), and tufA(MAE_42770, marked with orange) were all upregulated, which were essential regulatory factors for gene transcription and translation (Fig.3a).Meanwhile, hub genes (top10) were identif ied including 30S ribosomal proteins and 50S ribosomal proteins and marked with red series among the network (Fig.3b).
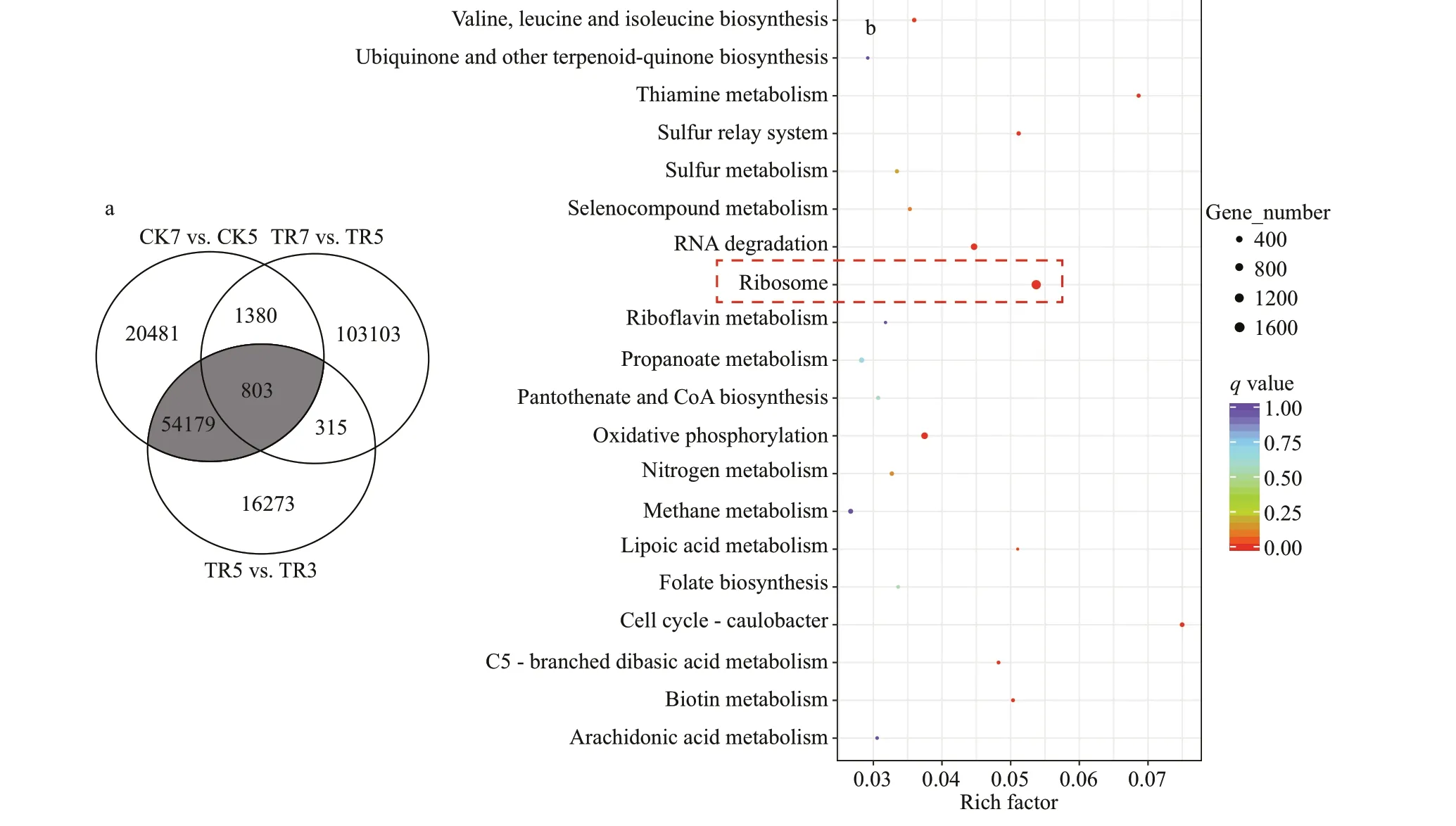
Fig.2 Gene expression and functional analysis
A module is a group of closely related proteins that act in concert to perform specif ic biological functions through the PPI network (Lin et al., 2015). There were 3 functional modules extracted from the PPI network (Fig.3c). Among them, module 1 that contained translation initiation factors and elongation factors was the largest functional module with a signif icant score (31 proteins, 409 interactions).KEGG enrichment analysis was conducted for proteins from module 1, showed that the ‘Ribosome’’was also the most enriched pathway (Table 1). This demonstrated that the core function of benthic bacteria-responsive unigenes was involved in ribosome inM.aeruginosa.
3.4 Expression analysis of Chl a synthesis related unigenes
In total of 5 enzymes that catalyzed the biosynthesis of Chlawere matched through the Swiss-Prot database,including magnesium chelatase (106 unigenes),magnesium-protoporphyrin O-methyltransferase(30 unigenes), anaerobic magnesium-protoporphyrin IX monomethyl ester cyclase (37 unigenes), divinyl chlorophyllide a 8-vinyl-reductase, chloroplastic(40 unigenes), and light-dependent protochlorophyllide reductase (17 unigenes, Fig.4a). Results from the hierarchical cluster analysis showed that Chla-related unigenes were more likely to high express in sample CK7 and TR5. These two samples were clustered together with similar expression patterns of unigenes.It may be contributed by synthetic activities of proteins from the ribosomes (Fig.4b).

Fig.3 Protein to protein analysis of benthic bacteria responsive unigenes
To be continued
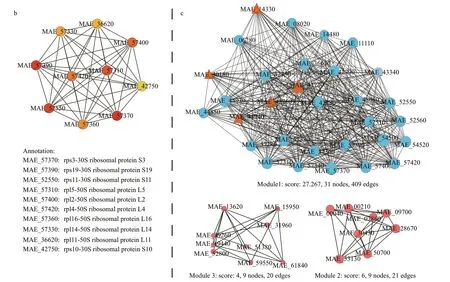
Fig.3 Continued
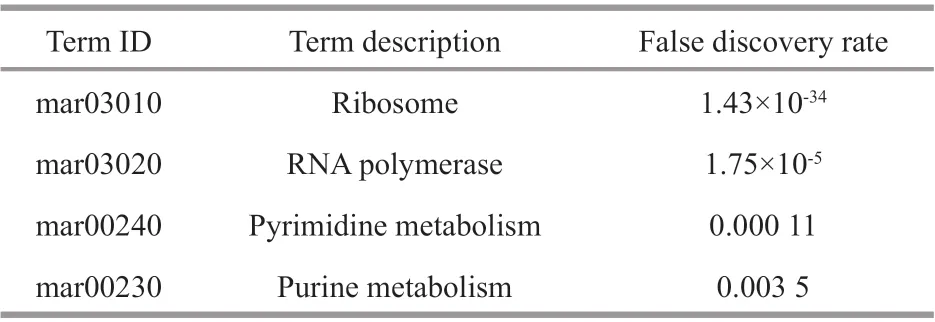
Table 1 KEGG enrichment analysis for unigenes from the module 1
4 DISCUSSION
The cyanobacteriumM.aeruginosais a dominant species in many lakes, which tends to aggregate at the surface to form bloom on the water (Wilson et al.,2005). They were hibernated on lake sediments in winter, due to poor conditions like limited light and low temperature (Cirés et al., 2013). Bacteria constitute a crucial component of aquatic ecosystems,and the close interactions between theMicrocystisbloom have received considerable attention (Grossart et al., 2006; Parveen et al., 2013; Landa et al., 2016).Buchan et al. (2014) indicated that Flavobacteria and Proteobacteria were dominant bacterial taxa that related toMicrocystisblooms; Xu et al. (2018)suggested thatEnterobacteraerogenescould enhanceM.aeruginosarecovery, and increase the expression of nicotinamide adenine dinucleotide (NADH)synthase and polysaccharide export-related genes. In our previous study, three benthic bacteria that promoted the recovery ofM.aeruginosawere isolated from the sediment of Chongtian Lake (Zou et al.,2018). In this study, transcriptional response ofM.aeruginosato the recruitment promoting-benthic bacteria at the early stage of recovery was further investigated, underling the important role of benthic bacteria on theMicrocystisbloom.
Ribosome was the ancient and highly conserved machinery that translates the genetic code into functional proteins. Studies suggested that the ribosomes of cyanobacteria would be in an inactive state during the stationary phase and in response to stress (ribosome hibernation), which was one prominent molecular strategy for post-stress survival(Wilson and Nierhaus, 2007; Trösch and Willmund,2019). Hence, the activated ribosome was an essential part during the recovery ofMicrocystis. Here, a set of unigenes were identif ied to be expressed early during the recovery ofM.aeruginosa, when in response to benthic bacteria. Results from the KEGG enrichment analysis showed that the function of these unigenes was related to ribosome. During the translation ofM.aeruginosacell, the mRNA f irst binds to the small ribosomal subunit (30S subunit), forming a complex together with the three initiation factors IF-1, IF-2,and IF-3, then the initiation complex interacts with the large ribosomal subunit (50S subunit) to assemble the full 70S ribosome. The peptide chain is continuously extended by the action of elongation factors lepA, fusA, and tufA. (Green and Noller,1997). In this study, the hub genes of the network mediated by benthic bacteria-responsive unigenes all were ribosomal proteins of the 30S and 50S.Meanwhile, regulatory factors including translation initiation factors (IF-2 and IF-3) and elongation factors (lepA, fusA, and tufA) were up-regulated in the largest functional module that related to the ribosome. These demonstrated that the benthic bacteria had a positive inf luence on activating the ribosome during the early recovery stage ofM.aeruginosa.
An active ribosome could provide abundant enzymes that catalyze various cellular processes. In this study, expression prof iles of unigenes encoding the enzymes that catalyze the biosynthesis of Chlawas characterized by the heat map. These unigenes were more likely to be highly expressed in sample CK7 and TR5, which may be indirectly inf luenced by the activated ribosome inM.aeruginosa. This study underlined the role of benthic bacteria in the recovery ofM.aeruginosa, and provided new thinking for understanding the regulation ofMicrocystisbloom.
5 CONCLUSION
In the presented study, we characterized the transcriptional response ofM.aeruginosato the recruitment promoting-benthic bacteria. There were 54 982 unigenes identif ied to be expressed earlier in response to benthic bacteria based on the expression level analysis. Results from the PPI analysis showed that hub genes of benthic bacteria-responsive unigenes mediated network were ribosomal proteins of 30S and 50S, and the most signif icant functional module of the network was also related to the ribosome.Unigenes encoding the translation initiation factors(IF-2, IF-3) and elongation factors (lepA, fusA, and tufA) were both up-regulated triggered by benthic bacteria. These indicated that the benthic bacteria had a positive inf luence on activating the ribosome during the early recovery stage ofM.aeruginosa.
6 DATA AVAILABILITY STATEMENT
The datasets generated during the current study are available from the corresponding author on reasonable request.
 Journal of Oceanology and Limnology2022年1期
Journal of Oceanology and Limnology2022年1期
- Journal of Oceanology and Limnology的其它文章
- The adjoint-based Two Oceans One Sea State Estimate(TOOSSE)*
- Structure and formation of the South Yellow Sea water mass in the spring of 2007*
- Lagrangian eddies in the Northwestern Pacif ic Ocean*
- Seasonal variability in dissolved oxygen in the Bohai Sea,China*
- In-situ experiments reveal mineralization details of porphyry copper deposits
- Chemical composition and Pb(II) binding of dissolved organic matter in a hypersaline lake in China*