
色素性痒疹1 例
2022-03-04 09:06石晶顾安康高琴
中国中西医结合皮肤性病学杂志 2022年1期
石晶,顾安康,高琴
(天津市中医药研究院附属医院,天津 300120)
1 病例资料
患者女,34 岁,躯干皮疹伴痒2 个月于2020 年5 月2 日来我院就诊。2 个月前无明显诱因自躯干出现红色丘疹,红斑,伴瘙痒明显,后皮疹逐渐加重,面积扩大,进而融合成片,部分可自行消退,遗留褐色色素沉着。皮疹时轻时重,反复发作,曾于我院门诊诊为“湿疹”,予以外用激素药膏及口服抗过敏药物治疗,未见明显好转。既往体健,个人史及家族史无特殊。
体格检查:系统检查未见异常。专科情况:前胸可见大片网状分布褐色色素沉着斑,融合成片,表面未见鳞屑,未见毛细血管扩张,触之未及萎缩,见图1。
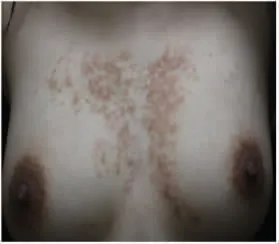
图1 患者前胸部皮疹
实验室检查:血常规和尿常规未见异常,皮疹真菌检查阴性。皮疹组织病理活检示:表皮轻度角化过度,棘细胞间水肿,可见散在角化不良细胞,表皮内有中性粒细胞移入;真皮乳头水肿,毛细血管扩张,血管周围以淋巴细胞为主的炎性细胞浸润,可见噬色素细胞,见图2。直接免疫荧光检查无明显特异性。
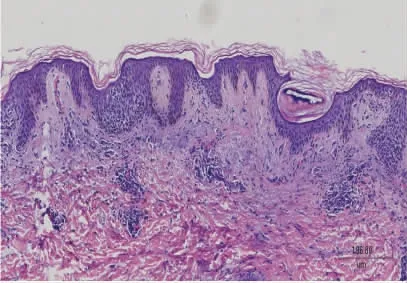
图2 患者皮损组织病理(HE 染色×10)
诊断:色素性痒疹(Prurigo pigmentosa,PP)。
治疗:口服盐酸米诺环素胶囊50 mg/次,2 次/d,治疗2 周后,瘙痒明显缓解,色素沉着斑较前明显变浅。随访1 个月,皮疹未复发。
2 讨论
PP 是一种少见的炎症性瘙痒性皮肤病,最早由日本医生Nagashima 等[1]于1971 年报道。该病多见于年轻女性(男女比例为1∶2),临床表现通常为躯干的红色丘疹、丘疱疹、水疱,后逐渐融合,最终遗留特征性的网状色素沉着,一般不累及黏膜。亦有报道发生于面部[2]。通常瘙痒明显,反复发作,多在春夏季,复发多局限于色素沉着区。本病病因及发病机制尚未明确,一些与代谢(糖尿病,酮症[3])、激素(妊娠)、物理因素(皮肤摩擦,创伤,光,热)、接触过敏源相关的因素被认为在发病中起作用。另有报道称幽门螺杆菌[4]感染亦是该病加重的主要因素。其中对酮症引起的代谢紊乱与该病发生的关联性研究较多,考虑酮症引起的中性粒细胞介导的皮肤炎性反应是该病的发病机制。目前全世界对PP 的报道已超过300 例[5],认为本病少被诊断可能是临床医生对其缺乏认知的原因。
本病组织病理在疾病发展的不同时期表现不同的特征:早期皮损一般出现于48 h 以内,可见真皮浅层血管周围中性粒细胞浸润,后中性粒细胞可逐渐移入水肿的真皮乳头及表皮,可伴棘层海绵水肿、气球样变性、角质形成细胞坏死;2 d 内充分发展后可见真皮浅层有程度不等的以淋巴细胞和嗜酸性粒细胞为主的浸润,可呈苔藓样,可因棘层水肿形成表皮内疱或表皮下疱。最终可见真皮浅层以淋巴细胞为主的炎性浸润,并可见噬色素细胞。最后阶段的组织病理与其他疾病导致的炎性反应后色素沉着难以区分,使得诊断困难。
本病常需与湿疹、融合性网状乳头瘤病等鉴别。湿疹皮疹形态多形性,对称分布,急性期渗出,迁延慢性浸润肥厚。常反复发作,瘙痒剧烈。组织病理急性期为海绵水肿性皮炎表现,慢性期呈慢性单纯性苔藓样表现;融合性网状乳头瘤病皮疹多见于胸部疣状或乳头瘤状丘疹,融合成网状,瘙痒常不明显,组织病理表皮呈乳头瘤状增生,真皮无炎性反应;本例患者反复发作的炎症性皮损及特征性网状色素沉着,组织病理学表皮轻度角化过度,棘细胞间水肿,可见散在角化不良细胞,表皮内有中性粒细胞移入;真皮乳头水肿,毛细血管扩张,血管周围以淋巴细胞为主的炎性细胞浸润,可见噬色素细胞。结合临床及病理符合该病表现,故诊断明确。
该病局部及系统应用糖皮质激素及抗组胺药治疗通常无效,其治疗方案主要为恢复正常饮食,改善摄入[3,6],并服用抑制中性粒细胞趋化的抗生素,如四环素类抗生素(多西环素[7-8]、米诺环素),或大环内酯类及氨苯砜。近期也有激光治疗有效的报道[9]。本例患者予以盐酸米诺环素50 mg/次,2 次/d,瘙痒明显缓解,色素沉着斑较前明显变浅。随访1 个月,皮疹未复发。该患者治疗效果较好,考虑与患者病程短,治疗及时有关。故对PP 早期诊断及合理治疗有助于改善预后。
猜你喜欢
西部皮革(2022年19期)2022-11-20
昆钢科技(2022年1期)2022-04-19
发明与创新·中学生(2020年6期)2020-06-22
时代汽车(2019年14期)2019-10-21
小学教学参考(数学)(2019年7期)2019-08-07
家庭医药·快乐养生(2019年2期)2019-03-04
科学与财富(2018年24期)2018-08-24
凤凰生活(2016年4期)2016-04-07
右江医学(2014年1期)2014-03-22
疯狂英语·口语版(2013年8期)2013-08-19
