
多视角多注意力融合分子特征的药物-靶标亲和力预测
2022-02-26 06:58:56王润泽张月琴秦琪琦张泽华郭旭敏
计算机应用 2022年1期
王润泽,张月琴*,秦琪琦,张泽华,郭旭敏
(1.太原理工大学信息与计算机学院,太原 030600;2.山西青年职业学院计算机与信息工程系,太原 030032)
0 引言
药物研发中一个不可或缺的过程是为蛋白质目标靶位筛选可结合并能产生效用的药物化合物[1]。自2020 年新型冠状病毒在全世界范围出现以来,针对病毒的基因序列,快速发现可结合的药物化合物以加速疫苗研发已成为学者们关注的焦点[2-3]。传统的药物研发通过大量的体外实验,为目标蛋白质的靶位(或者基因)筛选候选配体物。但是随着相关数据库中分子及化合物数据量剧增,如仅PubChem[4]包含110 M 化合物,传统的目标靶位精准筛选候选药物方法、不仅产生高昂的研发成本,更使得药物研发周期增长,耽误相关疾病的治疗[5]。研究者们进而考虑计算机辅助药物研发。现有研究主要集中于基于3D 化合物结构的计算方法,简化输入的机器学习方法以及自动特征提取的深度学习方法。特别是基于深度学习预测药物-靶标相互作用(Drug-Target Interaction,DTI)和药物-靶标亲和力(Drug-Target Affinity,DTA),成为目前研究的热点。目前的研究虽能够自动提取到有效的生物特征,但致力于将分子-蛋白质单方面结构嵌入到特征空间,单一方面结构信息对于精确提取分子的特征存在缺失不完备性。Lin 等[6]将分子局部图结构和序列结构嵌入的特征向量直接拼接作为深度神经网络的输入以预测亲和力。但简单的融合方式导致无法捕捉到与蛋白质靶位链接相关性更高的药物特征,影响两者结合强度的预测。
首先,分子不同视角生物属性对最终特征嵌入有增益;其次,不同视角的分子结构数据形态不同,所以需要有针对性的特征嵌入方式;最后,通过融合加权的多视角分子特征,能够捕获对靶位链接相关性更高的特征。
由此,提出多视角多注意力融合分子特征的端到端深度学习方法Ma2DTA(Multi-aspect Multi-attention Drug-Target Affinity),综合学习分子特征表示执行DTA 预测任务。Ma2DTA 主要包含两个核心模块:多视角分子结构嵌入(Multi-aspect molecular structure embedding,Mas)和多注意力融合(Multi-attention feature fusion,Mat)。首先,Ma2DTA 将分子全局拓扑结构、原子关联关系、原子化合键顺序排列进行嵌入。鉴于分子图这类非欧空间数据以及卷积神经网络对于局部结构的建模能力,利用图卷积神经网络学习分子图上原子与邻域原子节点之间的关联关系特征[7],并且融入图节点层级的注意力网络提取分子全局拓扑结构特征。其次,提出注意力融合多视角特征,捕获药物结构中对靶位链接亲和力更高的部分。最后,不同于将预测药物-靶标相互作用作为二分类任务,预测药物-蛋白质亲和力分数输出表示链接结合强度的连续值,弱关系药物-靶标对被舍弃,以缩小候选药物空间,加速药物研发进程,如图1 所示。实验结果表明,基于注意力融合多视角分子特征能够有效地捕获到对目标靶位链接亲和力更高的分子结构特征。
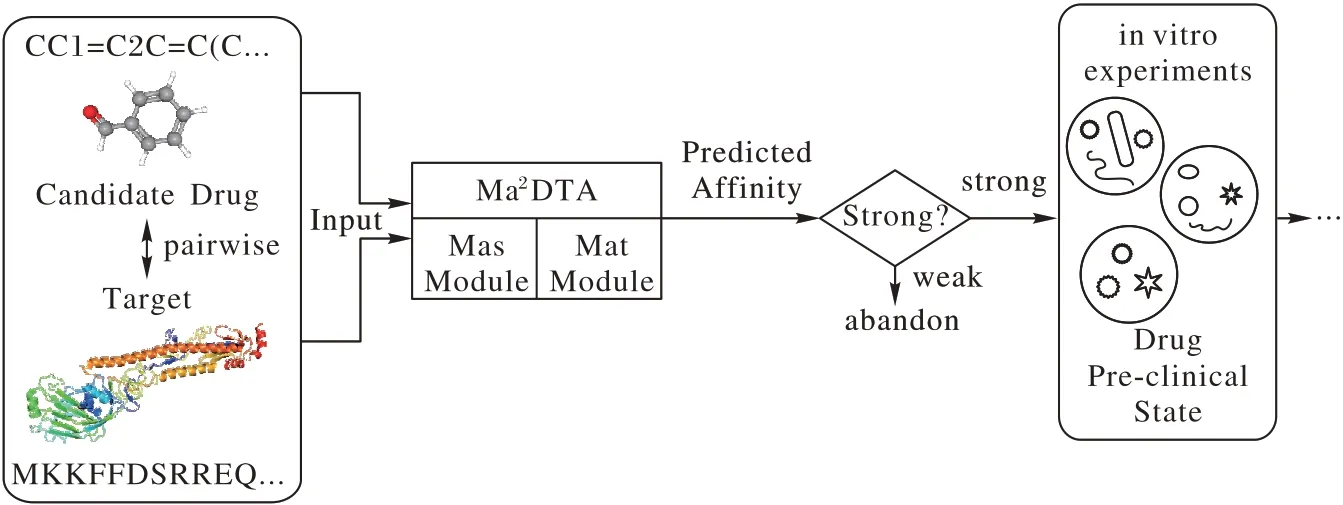
图1 药物研发阶段中的Ma2DTAFig.1 Ma2DTA in drug discovery stage
本文的主要工作概括如下:
1)针对分子多类型结构信息,提出注意力多视角分子特征融合方法,使得分子嵌入特征表示不同生物性质的信息融合增益。
2)提出一种分子特征层级的多注意力融合策略Mat,根据目标蛋白质,为每类分子结构特征附加亲和力权重,捕捉到对目标靶位亲和力更高的结构。
3)针对不同类型的分子信息具有不同的生物性质和形态,Mas 模块可通过不同嵌入的融合,学习代表其特有生物属性的特征向量。在两大数据集上实验表明Ma2DTA 的预测性能优于当前基准方法。
1 相关工作
早期代表性方法分子拼接[8]通过分析分子和蛋白质链接后的混合3D 结构,揭示分子链接靶位的机制。但由于3D 结构数据存在不易获取的局限性,研究人员考虑形式简易且生物属性表达性强的结构数据如生物实体之间的相似矩阵、关系矩阵和特征向量作为模型输入,利用机器学习方法预测药物-靶标关系。Perlman 等[9]提出集成药物-药物、基因-基因相似性度量矩阵,结合逻辑回归预测药物-蛋白质相互关系。Wang 等[10]提出一种关系矩阵补全方法融入药物相似性和蛋白质相似性作为对偶拉普拉斯正则项提升模型预测性能。He 等[11]从特征工程的角度提取药物-蛋白质对的相似性特征,引入梯度增强机来预测量化水平的药物-靶标亲和力。虽然这些方法解决了数据输入问题,并提高了预测的准确率,但是不可避免存在以下问题:生物数据获取困难、标签数据稀少、关系矩阵容量太大导致模型无法匹配、特征工程和专家经验引起的信息丢失和有偏。
深度学习能够在大规模数据中自动提取局部结构特征,受到研究者的广泛关注。深度学习助力药物研发也取得突破性进展[12-13]。特别地,DeepMind 在预测蛋白质3D 结构中取得不俗的成绩,科学家表示其有望改变生物学[14]。基于深度学习的方法在预测药物-蛋白质关系研究上也取得了进展。Wen 等[15]结合深度信念网络,从预训练和监督微调的角度预测 两者关 系。Öztürk 等[16]提出将 化合物SMILES(Simplified Molecular-Input Line-Entry System)序列和氨基酸序列作为输入,利用卷积神经网络(Convolutional Neural Network,CNN)分别提取局部关系特征。Karimi 等[17]结合注意力机制组合循环神经网络(Recurrent Neural Network,RNN)和CNN 从局部和全局联合学习序列特征预测亲和力。除此之外,另外一类方法利用分子结构本身存在的特性——可以自然地建模为图,图上的节点和边分别表示分子中的原子和化学键[18]。Gao 等[19]在分子图上定义卷积核实现原子节点与邻域间信息传播以挖掘分子图局部结构。Nguyen等[20]在分子图上执行不同类型的图卷积算法捕获分子图拓扑结构特征,提高模型预测亲和力能力。Li 等[21]同时编码分子图上节点和边的特征,结合原子与残基非共价关系指导亲和力预测。无论是基于序列结构还是图拓扑输入,上述方法致力于将分子-蛋白质单方面结构嵌入到特征向量空间,但单一方面的结构信息对于精确提取分子的特征存在缺失不完备性。
不同于上述方法,本文综合考虑分子多视角结构信息,通过注意力融合多视角分子结构特征,捕获对靶位链接相关性更高的部分分子结构。实验结果表明,相较于现有方法,Ma2DTA 在药物-靶标亲和力预测上能够达到更优的性能。
2 问题描述
本文的任务是融合多视角分子结构信息——分子拓扑空间、原子关联关系、原子化学键顺序排列结构,得到分子的综合特征向量表示,根据给定蛋白质预测药物-靶标亲和力。
首先,模型的原始输入数据为药物SMILES 串和氨基酸序列,具体表示如图2(a)输入部分所示。

图2 Ma2DTA的框架Fig.2 Framework of Ma2DTA
其次,利用化学信息工具RDKit[22]将输入的SMILES 串转换为分子图Gm={V,E},其中vi∈V(i=1,2,…,Natom)表示分子中的第i个原子节点,ei,j∈E(i,j∈{1,2,…,Na})表示分子图中的第i个原子与第j个原子之间的化学键。使用{S1,S2,…,SNs}与{T1,T2,…,TNt}分别表示分子SMILES 串和蛋白质序列的标识符集合,Ns和Nt分别代表两者序列各自的长度。由于不同的分子结构信息包含不同的生物属性信息,具有不同的性质,所以针对不同的分子结构需要不同的嵌入方法。因此需要学习的原子关联关系结构嵌入函数为fv_a(·):

其中hloc为原子关联关系嵌入特征向量。分子全局拓扑结构嵌入函数为fv_s(·):

其中hsuper为学习的分子全局拓扑结构特征向量。vsuper表示在分子图外定义的存储全局图拓扑特征的节点[22]。原子与化学键排列结构嵌入函数为fv_sim(·):

其中hsmi表示原子与化学键排列结构嵌入特征向量。针对蛋白质结构,需要学习的表征函数为fpro(·):

其中hpro为学习的蛋白质结构特征。得到3 个视角的分子结构特征后,通过注意力函数为多视角分子结构嵌入向量附加权重融合:
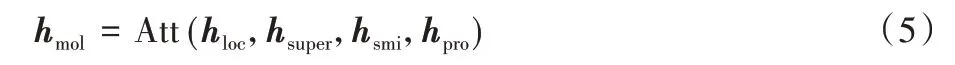
其中:hmol为嵌入的分子最终特征表示,Att(·)表示注意力融合函数。则整体模型的优化可定义为:
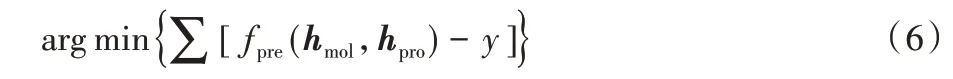
其中:fpre(·)表示亲和力回归预测函数,y为真实的亲和力分数值。
3 模型提出
本章分别介绍多视角分子结构嵌入模块Mas、蛋白质结构编码模块、多注意力分子特征融合模块Mat 和药物-靶标亲和力回归预测模块。Ma2DTA 整体框架如图2 所示:图2(a)描述Ma2DTA 从数据输入到亲和力输出的框架,以药物的SMILES 串和蛋白质的氨基酸序列作为模型输入,通过Mas 模块将三种类型的分子结构和蛋白质序列结构分别嵌入为代表各自生物属性的特征向量;图2(b)表示Mas 模块中分子全局拓扑结构和原子关联关系结构的嵌入过程;图2(c)展示原子化学键顺序排列结构的特征学习过程;图2(d)表示蛋白质序列结构的特征嵌入。在Mat 模块中,根据计算的分子结构相对于靶蛋白的重要性系数,对三类分子特征向量加权,然后融合得到给定药物的最终特征表示。最后,将药物与蛋白质特征向量拼接后利用多层神经网络拟合输出亲和力分数。
3.1 多视角分子结构嵌入模块Mas
多视角分子结构嵌入模块针对不同生物属性的分子结构采用不同的表征策略。将5 类原子属性——原子标签、原子节点的度、氢原子的总数、原子的隐含值、原子是否为芳香族采用one-hot 分别编码为向量hlab、hdeg、hH、hhid、haro,则每个原子节点的初始化特征表示为:

其中||表示向量级联。
针对原子关联关系结构,优化嵌入函数fv_a(·)以学习分子中每一个原子与邻域原子节点消息传播的特性,最终得到关联特征hloc。采用图卷积神经网络(Graph Convolutional Neural Network,GCN)[23]在频谱空间上对分子图执行卷积运算。式(8)定义了每一层图卷积网络的计算过程:

其中:A∈为分子图邻接矩阵,I为大小相同的单位矩阵,D是图对角度矩阵,X和Θ分别表示分子特征矩阵和可学习的参数矩阵,Z∈表示每一次图卷积后的中间隐含特征矩阵,dl为第l层原子的特征维度,ReLU(·)为非线性激活函数。最后,通过图全局池化层得到分子的最终关联关系特征hloc。
针对分子全局拓扑结构,则需要通过优化fv_s(·)以嵌入全局图拓扑结构得到特征hsuper。此处通过图节点层级的注意力捕获每一个原子对分子全局结构特征的贡献。第一,累加每个原子特征初始化全局节点vsuper;第二,在计算图节点层级注意力过程中融入多头注意力机制集成多个空间的注意力信息以提升对注意力权重的学习。在注意力系数的计算中首先利用式(9)计算每一个原子节点对超节点vsuper贡献系数:

针对原子和化学键顺序排列结构,优化嵌入函数fv_sim(·)以提取原子-化学键序列局部结构特征hsmi。首先结合标签嵌入和随机初始化将SMILES 标识符集合{S1,S2,…,}转换为语义向量组[s1,s2,…,],si为具有固定维度的特定语义向量。其次采用CNN 学习序列中每一个标识符的局部上下文信息,即捕获局部原子与化学键连接关系,式(14)定义卷积的特征捕获过程:

3.2 蛋白质结构编码模块
对于模型输入的蛋白质序列,同样通过优化表征函数fpro(·) 捕获氨基酸顺序排列特征hpro。类似于上文中对SMILES 序列编码,使用标签嵌入和随机初始化将氨基酸序列的标识符集合{T1,T2,…,}编码为向量组[t1,t2,…,]。采用CNN 提取蛋白质序列中局部氨基酸顺序排列特征:

3.3 多注意力分子特征融合模块Mat
本模块提出一种多注意力融合方式,得到代表不同分子生物性质的信息融合增益。第一,不同类型的分子结构表示不同的生物属性,对最终的分子特征表示提供不同程度的信息增益;第二,多注意力融合能根据亲和力权重捕获分子上链接靶位更重要的特征;第三,粗略的融合方式无法突显对靶位亲和力更高的分子特征。基于此,本文提出分子结构层级的注意力融合方法建模分子结构对蛋白质靶位链接相对重要的特征,提高模型预测亲和力的性能。Mat 模块优化注意力函数Att(·),融合多视角分子结构特征得到分子最终特征表示hmol。给定Mas 模块嵌入的分子特征向量hloc、hsuper、hsmi,定义其对应的注意力权重分别为βloc、βsuper、βsmi。以βloc为例,其计算如式(16)、(17)所示:

其中:Wtrans,2表示分子结构层级的注意力参数矩阵;Wattn,l、Wattn,p分别代表原子关联关系特征和蛋白质结构特征相对应的线性特征变换矩阵。注意力权重βsuper、βsmi以相同的计算过程得到。最后,对不同视角下的分子特征根据各自的注意力系数加权,经过非线性聚合得到分子最终特征表示hmol,如式(18)所示:
《中华人民共和国印花税暂行条例施行细则》[9]中明确规定:国家指定的收购部门与村民委员会、农民个人书立的农副产品收购合同免纳印花税。

其中Wmol为分子最终嵌入特征的线性变换矩阵。
3.4 药物-靶标亲和力回归预测
本阶段利用回归预测函数fpre(·)完成药物-靶标亲和力预测。根据药物-蛋白质对,其中h=[hmol||hpro]表示hmol与hpro的拼接,经过L层深度特征变换输出药物-蛋白质亲和力分数score:

4 实验与结果分析
在浪潮异构机群GPU:12 *32 GB Tesla V100s,内存640 GB DDR2 进行实验,验证Ma2DTA 的亲和力预测性能。
4.1 数据集
在Davis[24]和KIBA[25]数据集上进行实验。Davis 数据集包含激酶家族及其抑制剂的蛋白质样品,以及相应的解离常数(dissociation constant)值Kd,本文使用通用方法将Davis 数据集中的Kd值转换到log 空间以保证数值的平稳性,如式(20):

KIBA 数据集整合抑制剂生物活性的各种来源,通过应用其统计信息优化抑制常数(inhibition constant)Ki、Kd和半抑制浓度(half-maximal inhibitory concentration)IC50 之间的一致性。表1 给出了两个数据集上的数据统计。

表1 实验数据集统计信息Tab.1 Statistics of experimental datasets
4.2 评价指标

其中:bi是较大亲和力δi的预测值,bj是较小亲和力δj的预测值;Z是一个标准化常数。h(x)表示梯函数:

MSE 量化模型预测值与数据真实值之间的差异性,如式(23)所示:

其中yi表示真实值。MSE 越小,模型的预测效果越好。评估模型对数据的拟合能力。越大,表明模型拟合程度越好,如式(24):

其中r2和分别代表有截距和无截距时的平方相关系数。
4.3 实验设置
为验证Ma2DTA 的亲和力预测性能,分别将其同以下四个基准深度学习模型比较。
1)DeepDTA[16]:端到端的深度学习系统,从分子和蛋白质序列结构的角度提取特征。
2)AttentionDTA[27]:根据模型学习的SMILES 序列和氨基酸序列片段之间的权重预测可能性更高的链接位置。
3)GANsDTA[28]:构建一个半监督学习系统,从药物-蛋白质无标签的角度,采用生成对抗网络提取药物与蛋白质特征。
4)GraphDTA[20]:表征分子图拓扑结构和蛋白质氨基酸序列结构预测药物-蛋白质亲和力。
由于SMILES 序列和氨基酸序列长度不等,为保证效果对比的公平性,实验中对SMILES 序列和蛋白质序列分别设置固定长度为85 和1 000,大于固定值的部分被截断,小于固定值的部分用0 补充。实验表明,多视角分子结构融入和多注意力融合均有助于药物-靶标亲和力预测性能的提升。
4.4 结果分析
在Davis 和KIBA 数据集上执行实验分析,将本文方法与当前基准方法进行比较,分别考虑训练集的分割比率的影响,多视角特征融入和多注意力融合的有效性,以及对蛋白质序列卷积的层数和分子图上的池化方式的影响。
4.4.1 训练比率的影响
为验证训练集的所占比率对模型性能的影响,实验中将两个数据集分割为训练集和测试集,分别设置4 个不同的训练比率——80%、60%、40%、20%,测试集比率对应为20%、40%、60%、80%。图3 展示在两个数据集上执行不同的数据分割对Ma2DTA 性能的影响。当训练比率设置为80%时,Ma2DTA 在所有指标上达到最优。
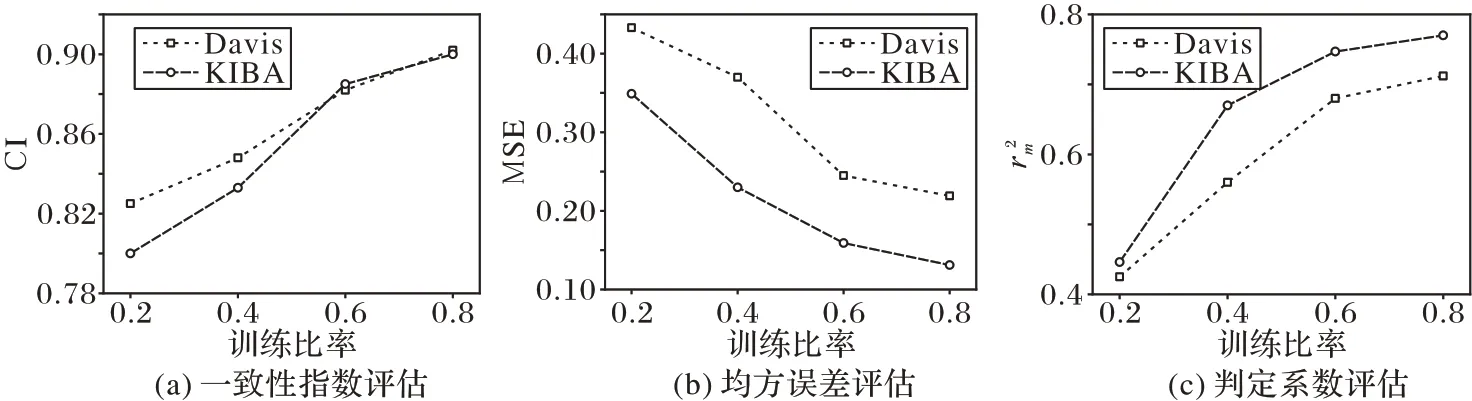
图3 两个数据集上训练比率对Ma2DTA性能的影响Fig.3 Influence of training ratio on Ma2DTA performance on two datasets
4.4.2 与基准方法相比
如图4 所示,在Davis 和KIBA 数据集上,将本文方法与基准深度学习方法分别作了比较。首先,基于图数据输入的方法(GraphDTA)相较于基于序列结构(DeepDTA,GANsDTA等)的方法,在CI 和MSE 上有明显的提升,表明分子天然地可建模为分子图的特性,可以有效地表达出原子与原子之间的关联关系以及拓扑空间携带的生物属性信息。其次,无论是 在Davis 还 是KIBA 数据集 上,Ma2DTA 在CI 指标上突破0.90,达到高准确度。在Davis 数据集上,Ma2DTA 在MSE 值上,比GraphDTA 降低接近5%,其比最好的基准方法AttentionDTA 提高了7%。在KIBA 数据集上,Ma2DTA 比基准方法中效果最明显的GraphDTA,MSE 降低6%,而比GraphDTA 大幅度提升接近10%,相较于在这个指标上效果显著的AttentionDTA 提高4%。实验性能比较如图4 所示,Ma2DTA 均优于基准方法,一方面,表明融入三个不同方面的结构能够使得分子最终特征表示包含更丰富的生物属性信息。另一方面,使用注意力机制从分子特征层面上融合,根据权重选择对目标靶位亲密度更高的部分结构执行融合,避免造成次优预测。
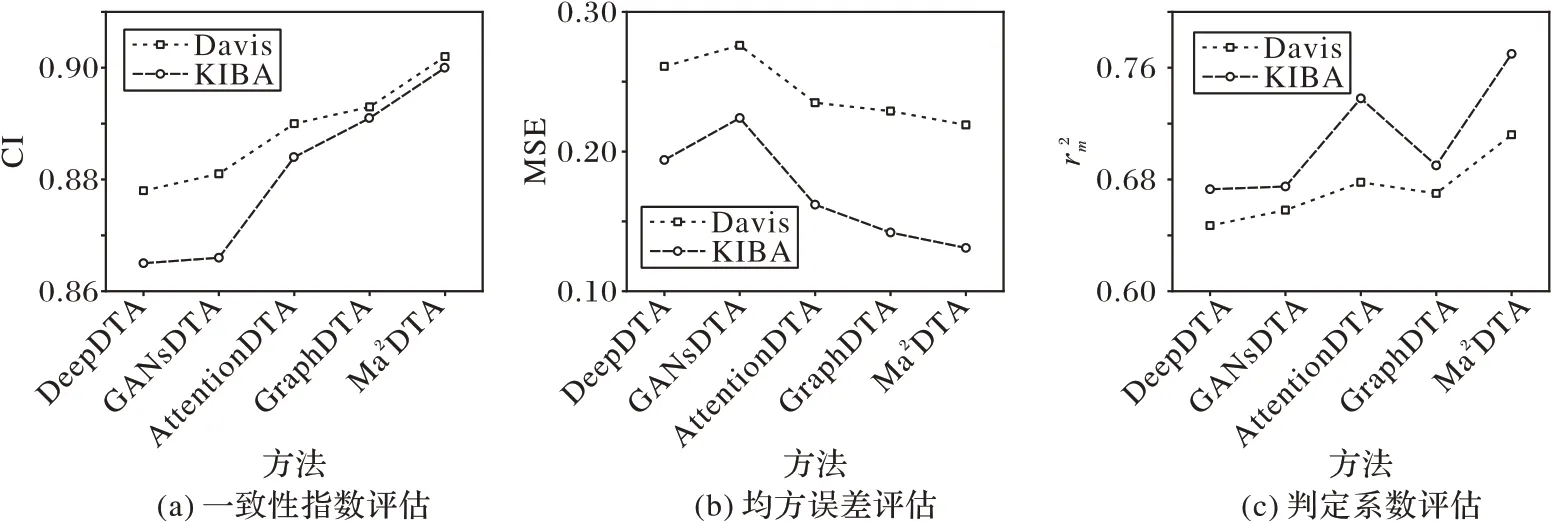
图4 两个数据集上所提方法与基准方法的比较Fig.4 Comparison of the proposed method with baseline methods on two datasets
4.4.3 多视角分子特征融合的有效性分析
为验证多视角分子特征融合的有效性,本文在Davis 数据集上分别比较一种结构、两种结构和三种结构的分子特征融合对预测性能的影响。在实验中,对涉及的序列结构均采用CNN 提取特征,对涉及的分子图采用GCN 表征。比较结果如图5 所示,MSE 随着特征融合数量的增加呈下降趋势,而CI 和则呈上升趋势,说明原子关联关系结构、分子图全局拓扑、原子化学键顺序排列有益于整体分子结构的表征学习,并且从多个视角能够综合地分析分子结构,有助于提高预测药物-蛋白质亲和力的效果。特别是Ma2DTA 除了在MSE 和CI 评估上均明显达到最优,相较于单一序列结构提取,MSE 降低16%,提高10%,CI 提高超过3%;而相较于单一局部分子图结构提取,MSE 降低了11%,提高6%,CI 明显提高接近3%。综上所述,Ma2DTA 采取多视角分子特征融合策略能够对分子最终特征表示产生信息增益。

图5 多视角分子结构融合的有效性Fig.5 Effectiveness of multi-aspect molecular structure fusion
4.4.4 多注意力融合的有效性分析
为验证提出的注意力融合方式有效性,分别比较多注意力融合、均值化、累加和,以及特征向量级联对预测结果的影响。实验执行过程中,为了仅考虑融合方式的影响,对于上述4 种融合方式,均采取相同的参数选择。如图6 所示,多注意力融合多视角分子特征,均优于其他的融合方式,表明基于多注意力机制的融合能够从分子结构层面,根据不同分子结构对目标蛋白质结构的权重系数,捕捉到对目标靶位亲密度更高的部分结构,使得分子最终嵌入特征表示不同生物性质的信息增益,从而提高药物-蛋白质亲和力预测性能。
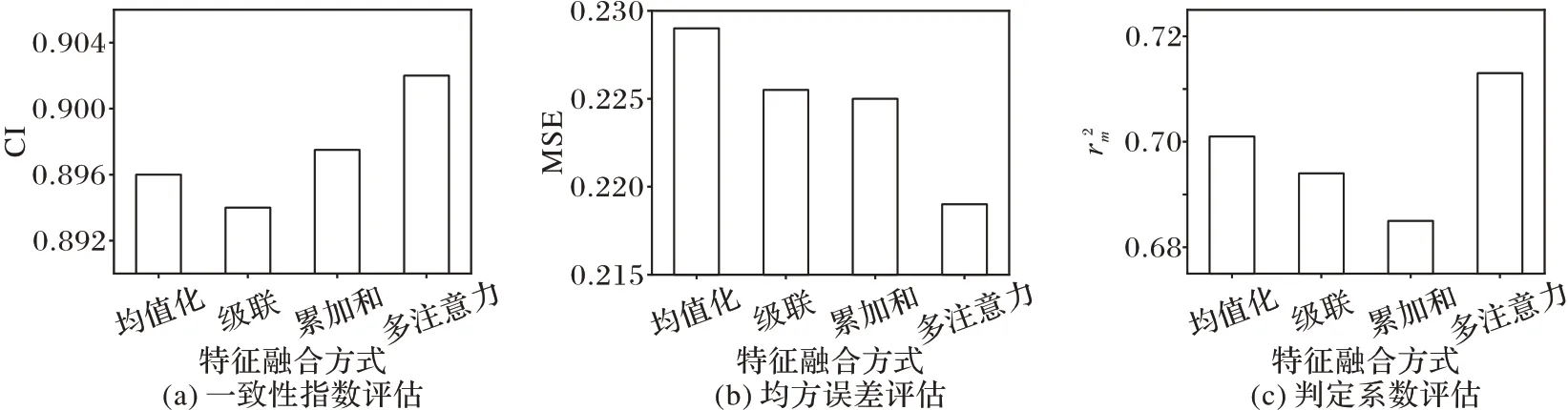
图6 注意力融合的有效性Fig.6 Effectiveness of attention fusion
4.4.5 重要参数选择
为考虑重要参数的选择对于模型预测性能的影响,在Davis 数据集上通过实验分别比较针对蛋白质序列结构的CNN 层数和针对分子图的全局池化方式。图7(a)给出当CNN 层数设置为1、2、3 时,三个评价指标的变化。图7(b)表示原子关联关系结构嵌入过程选用全局累加(sum)、最大(max)、平均池化(mean)时,对模型预测性能的影响。当针对蛋白质序列的卷积层数设置为3 层时,模型表现出最佳性能。当针对分子图使用全局最大池化时,由于映射出分子最突出显著的局部特征,相较于其他两种池化方式,性能最佳。
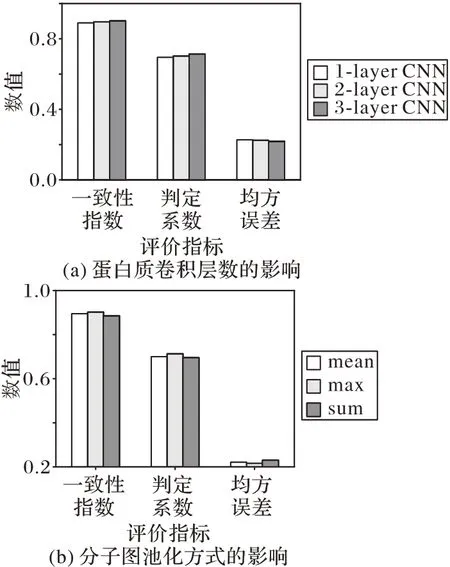
图7 蛋白质卷积层和分子图池化方式对性能的影响Fig.7 Influence of protein convolutional layers and molecular map pooling methods on performance
5 结语
本文提出了一种多视角注意力融合分子特征的药物-蛋白质亲和力预测方法Ma2DTA,分别从三个视角——原子关联关系、分子全局拓扑、原子化学键顺序排列提取不同的结构特征(Mas),通过分子特征层级的注意力融合获取每一类对目标靶位亲密度更高的部分结构(Mat),保留对分子最终特征表示最有益的信息。实验结果表明Ma2DTA 在预测药物-靶标亲和力任务上具有良好的性能。
未来的工作将进一步尝试多样化的图神经网络方法表征分子图,并且考虑不同分子视角数量对预测的影响,以及更深层次地考虑氨基酸和原子之间的相互作用。
猜你喜欢
中学化学(2024年5期)2024-07-08 09:24:57
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:44
新闻传播(2018年11期)2018-08-29 08:15:30
新闻传播(2018年13期)2018-08-29 01:06:52
中学化学(2016年10期)2017-01-07 08:37:06
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01 04:06:40
新闻传播(2016年9期)2016-09-26 12:20:34
