
LHCGR 基因突变致家族性男性性早熟2 例报告及文献复习
2022-02-19 08:54何亲羽陈立芬张雪蕾董治亚
诊断学(理论与实践) 2022年5期
何亲羽,王 伟,陈立芬,张雪蕾,董治亚
(1.上海交通大学医学院附属瑞金医院儿内科,上海 200025;2.上海交通大学医学院临床医学系,上海 200025)
家族性男性性早熟(familial male-limited precious Puberty,FMPP),又称为家族性高睾酮血症或睾酮毒症,是一种常染色体显性遗传性疾病,也是男性性早熟的罕见病因之一[1]。目前证实,该疾病是由于黄体生成素/人绒毛膜促性腺激素受体(luteinizing hormone/choriogonadotropin receptor,LHCGR)基因发生功能获得性突变,黄体生成素(luteinizing hormone,LH)受体功能增加,呈现自主激活状态,从而持续性发挥作用,使睾酮分泌增加。患病男童多在4 岁前因血清睾酮水平升高而出现外周性性早熟症状,包括生长加速明显、阴茎增大可伴有勃起、骨龄提前等,部分患者还伴有攻击性增加等行为问题,也可进展为中枢性性早熟(central precocious puberty,CPP)。本研究收集2 例就诊于本院且经临床诊断及基因测序确诊为LHCGR 基因突变所致性早熟的患者,结合文献复习,总结FMPP 的临床特征以及基因突变的相关特点,为男性性早熟病因诊断及FMPP 治疗随访提供参考。
资料与方法
一、研究对象
本研究纳入2 例我院儿科诊治的LHCGR 基因突变患者,收集其完整病史资料、诊断、治疗及随访经过。本研究获得上海交通大学医学院附属瑞金医院医学伦理委员会批准[(2021)临伦审第(16)号],患者监护人签署了知情同意书。
二、方法
1.实验室检查及影像学检查:予患者完善血常规、尿常规、糖脂代谢、肝肾功能、电解质、肿瘤指标检测;行促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)激发试验,并检测性激素、甲状腺功能以及肾上腺相关激素;行B 超(睾丸、腹部、肾上腺)、垂体核磁共振、骨龄片等影像学检查。
2.基因突变分析:经患者及其父母知情同意后,采集外周静脉血各4 mL,从白细胞中提取基因组DNA 进行高通量测序,并分析其致病基因。用Agilent SureSelect 方法进行外显子捕获,用Illumina测序平台进行高通量测序,测序数据经软件匹配分析,使用在线软件系统进行变异过筛及解释,候选变异经Sanger 测序验证。根据美国医学遗传学与基因组学学会(The America College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南进行评级。
3.治疗与随访:予患儿促GnRH 类似物(GnRH analogue,GnRHa)联合抗雄激素制剂治疗,经家属同意后加用芳香化酶抑制剂。随访患儿的生长速率、性征发育情况、骨龄及促性腺激素和性激素水平。
4.文献检索和资料分析:以“FMPP”“LHCGR”“家族性男性性早熟”“高睾酮血症” 为检索词通过PubMed 医学文献数据库检索国外文献,通过万方数据库检索国内文献,通过文献复习总结疾病临床及遗传特征。
结果
一、一般情况
1.病例1:患儿年龄为6 岁1 个月,因“外生殖器增大6 月个余” 就诊,5 岁7 个月龄时出现阴茎增大,勃起次数增多,伴面部痤疮,声音较前变粗,近半年身高增长5.5 cm。父亲身高159 cm,母亲身高162 cm,均否认青春发育提前病史。体格检查示,患儿身高137.1 cm,身高标准差积分为+4.22 SD,体重35 kg(>+3SD),体质量指数为18.6 kg/m2(+SD~+2SD),体型匀称,声音稍粗,面部可见明显痤疮,无胡须及喉结;双侧乳房发育Tanner Ⅱ期,阴毛发育Ⅱ期,阴茎7.7 cm×2.6 cm,双侧睾丸大小8 mL,未触及肿块;余查体未见异常。
2.病例2:患儿年龄为3 岁7 个月,因“自幼阴茎粗大”就诊。患儿出生后不久即发现阴茎较同龄儿粗大,当时未予特殊关注;自幼身高增长速度较快,冲动易怒,伴攻击行为。父亲身高173 cm,母亲身高163 cm,均否认青春发育提前病史。体格检查示,患儿身高110.5 cm,身高标准差积分为+2 SD,体重21.2 kg(>+2 SD),体质量指数为17.5 kg/m2(+SD~+2 SD),体型匀称,未变声。无痤疮,无皮肤牛奶咖啡斑;双侧乳房发育Tanner Ⅰ期,无阴毛生长,阴茎7.3 cm×2.4 cm,双侧睾丸大小6 mL,阴囊无明显色素加深。余查体未见异常。
二、实验室检查结果
2 例患儿的LHRH 激发试验结果示LH 峰值分别为7.28 mIU/mL 和4.96 mIU/mL,基础睾酮水平升高达2.49 ng/mL 和3.58 ng/mL,其余内分泌激素及血、尿常规和肝、肾功能、肿瘤指标等检查均正常。病例1 的骨龄为13 岁(见图1),病例2 的骨龄为5 岁(见图2)。腹部B 超、肾上腺B 超、睾丸B 超、垂体增强核磁共振均未见占位性病变及其他异常。

图1 病例1 骨龄约13 岁
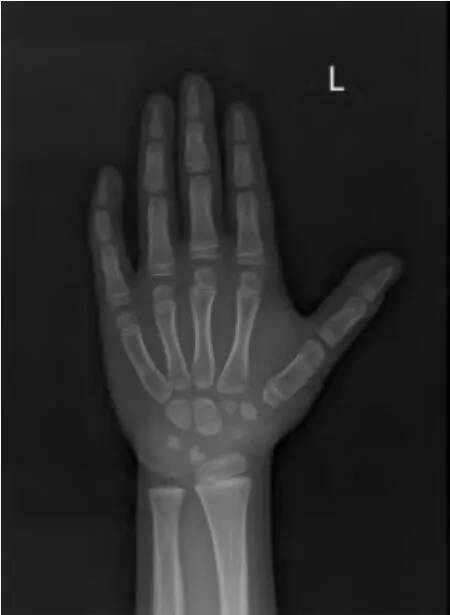
图2 病例2 骨龄约5 岁
根据患者典型的临床症状和体征,LHRH 激发试验结果提示下丘脑-垂体-性腺轴启动,睾酮水平升高,可临床诊断CPP,且影像学结果排除中枢系统病变、睾丸间质细胞肿瘤、先天性肾上腺皮质增生症等病因导致的性早熟(见表1)。

表1 2 例患者的实验室检查及影像学结果
三、基因测序结果
全外显子测序检测显示(见表2),病例1 LHCGR 基因第11 号外显子存在c.1756TCTdel 杂合缺失,TCT 这3 个碱基缺失导致第586 位氨基酸丝氨酸缺失,其父亲存在相同的碱基对缺失(见图3),母亲则正常,根据ACMG 变异分类指南归类为可能致病性变异。病例2 的LHCGR 基因第11 外显子则存c.A1723>C 杂合突变,导致第575位氨基酸残基由异亮氨酸突变为亮氨酸,该突变来自于母亲,父亲则正常(见图4),根据ACMG 指南评判为可能致病性变异。

表2 文献复习资料
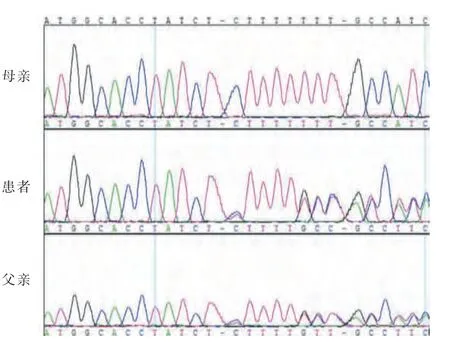
图3 病例1 及父母基因测序结果
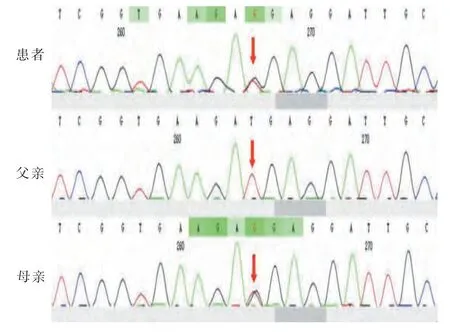
图4 病例2 及父母基因测序结果
四、治疗与随访
完善基因检测后,2 例患者均确诊为由LHCGR基因突变导致的FMPP 且并发CPP,予治疗和随访。
FMPP 的治疗常用药物有抗雄激素制剂、芳香化酶抑制剂以及P450 酶抑制剂等[2]。当并发CPP时,可加用GnRHa 抑制性腺轴。
病例1 至我院就诊前,在外院接受GnRHa 治疗,剂量为3.75 mg 每月注射1次,共治疗半年,睾丸未见明显退缩,生长速率仍快,骨龄继续进展,睾酮水平未见明显下降。转诊我院来完善基因检测更正诊断后,予氟康唑6 mg/(kg·d)、来曲唑1.25 mg/d口服,并联合GnRHa 治疗,同时抑制性腺轴与外周睾酮作用。随访4 年间,患者临床症状改善,睾丸逐渐缩小,未出现药物相关副反应。
病例2 在我院明确诊断后,即开始治疗,初始治疗方案为GnRHa 3 mg 每月注射1次,联合螺内酯1 mg/(kg·d)口服。随访半年后,生长速率仍偏快且骨龄控制欠佳,与家属充分沟通并签署同意书后,加用阿那曲唑0.5 mg/d 口服。生长速率明显减缓,睾丸未在持续增大,冲动易怒较前改善,末次随访时骨龄基本控制。
五、文献复习
以“FMPP”“LHCGR”“家族性男性性早熟”“睾酮毒症”为检索词,在PubMed 共检索到29 篇相关病例报道,在万方数据库共检索到4 篇中文病例报道,将其中资料相对完整的35 例进行基因型及特点进行总结(见表2)。LHCGR 基因突变患者多在4 岁前即有性早熟表现,少数也可在5~8 岁起病。绝大多数病例都存在血清睾酮水平升高达成人水平,骨龄有不同程度的提前,这也符合男性外周性性早熟的特点,但睾酮水平高低及骨龄提前程度与基因型之间无相关性。
讨论
一、FMPP 与LHCGR 基因
FMPP 最早在1981 年由Schedewie 等[25]进行描述,而Shenker 等[26]于1993 年首次从8 个不同家族的多名FMPP 患者中均检测到LHCGR 基因突变,从而将FMPP 与LHCGR 基因突变关联起来。LHCGR 基因编码LH 受体蛋白,该蛋白可以同时与LH 及人绒毛膜促性腺激素结合。正常情况下,青春期下丘脑-垂体-性腺轴启动后,LH 与睾丸间质细胞上的LH 受体结合,进而激活腺苷酸环化酶,促使睾酮合成增加,使男性出现青春发育。LHCGR基因发生功能获得性突变后,LH 受体不需与LH结合而呈持续性激活,导致细胞内非LH 依赖性的腺苷酸环化酶增加,睾丸间质细胞持续分泌睾酮,血清中睾酮水平过度增高,导致男性性早熟发生。但此前报告的该突变基因的女性携带者并无特定表型,这可能是因为青春期前女孩卵巢中LH 受体表达降低或缺失,也有研究认为是因为雌激素的产生和卵泡发育需要LH 和卵泡刺激素共同参与[27]。故病例2 的母亲虽携带与患者相同的基因变异,但无性早熟病史,并能正常生育。
至今国内外共报道了18 种LHCGR 基因激活性突变类型可引起FMPP 的病例[28],也有少量新发突变引起散发性男性性早熟[23,29]。大部分突变位于11 号外显子,其中1624~1741 是热点突变区,位于LHCGR 第6 跨膜区,编码第571~581 位氨基酸。其中第578 位的天冬氨酸(Asp578-)是热点突变氨基酸残基,约55%的突变影响到该残基,可突变为酪氨酸、组氨酸、甘氨酸和谷氨酸[17-18,22,28-32]。Asp578-突变在美国FMPP 患者中约占90%,但在我国的病例报道中多为甲硫氨酸被苏氨酸取代(Met398Thr),提示不同人种之间的突变类型存在差异。LHCGR突变可来源于父系,也可来源于母系,部分新发突变,根据既往文献报道,突变来源与基因型之间无相关性。
二、本研究患者的突变类型与临床特征
本研究中,病例1 的基因变异为碱基TCT 缺失导致编码的丝氨酸缺失,突变位点发生在LH 受体的第6 跨膜区。采用SWISS MODEL 软件绘制蛋白质三维结构模拟图,可见突变型(见图5)相较野生型(见图6)α 折叠减少,导致LH 受体构象改变。该突变是未曾报道过的新LHCGR 基因变异类型,根据ACMG 变异分类指南归类为可能致病性突变,但该突变如何引起LH 受体激活仍需行进一步功能验证。

图5 LHCGR 突变型蛋白三维结构模式图

图6 LHCGR 野生型蛋白三维模式图
虽然突变形式不同,但该病例的临床表现与既往的FMPP 病例相比,除骨龄与实际年龄差值较大(两者差值为7 岁)之外,在发病年龄、主要临床表现以及激素水平等方面未表现出显著差异。本研究推测,骨龄明显进展的原因是初始治疗时未能识别LHCGR 基因突变,由睾丸产生过多的睾酮持续向雌激素转化,加速了骨骺的成熟。既往报道的病例中也有骨龄明显大于实际年龄患者,其基因突变的位点各不相同,所以目前认为,骨龄成熟度与突变类型没有明确的相关性。该患者的变异来源于父亲,虽然父亲未能提供性发育的具体情况,但其成年身高仅为159 cm(-2.25 SD),与大多数FMPP 患者的成年终身高水平一致[15,33-34],推测患者父亲在幼年期也存在雄激素增高使骨龄增速,从而导致终身高矮小。
病例2 的突变同样位于热点区域11 号外显子,为第1 723 位的碱基由腺嘌呤突变为胞嘧啶(c.1723A>C),使编码的氨基酸由异亮氨酸变为亮氨酸(p.Ile575Leu),该突变位点已由Laue 等[23]及杨海花等[14]报道过。有研究[35]证实,Ile575Leu 突变使第6 跨膜区末端的细胞可溶性增加,改变了细胞表面表达,增加了与配体的亲和力,且体外功能实验表明,在无激动剂存在的情况下,表达突变类型受体的细胞内的腺苷酸环化酶比野生型增加15~25 倍。该位点突变所致的临床表现与其他FMPP 类似,皆以生长加速、外生殖器增大、骨龄加速、性格改变等高雄激素引起的临床表现为主。因女性携带该基因变异无症状,类似于病例2 的突变来源于母亲,无阳性家族史可询,临床易诊断性早熟,但容易忽视病因诊断,造成漏诊,延误治疗时机。
三、本研究患者的诊断过程
早期FMPP 难识别,容易漏诊,本研究2 例患者就诊时均已并发CPP,提示相较于女性,男性CPP 患者多有病理因素所致[36-37],但对病因的精准诊断尚存在一定的困难。本研究的病例1 在外院诊断为中枢性性早熟(病因不明,特发性可能),按中枢性性早熟常规使用GnRHa 治疗,但骨龄进展未见减缓,生长加速仍明显,外生殖器发育也未被抑制。经GnRHa 治疗,病例1 的血清促性腺激素基础值已降至青春前期水平,但睾酮仍处于青春期水平,提示睾酮的分泌增加并不仅仅是由于性腺轴启动,可能存在特殊原因使睾丸间质细胞持续分泌睾酮。此外,病例1 的父亲具有成年身高矮小的表现,具有可疑家族史,故考虑可能为继发性CPP,需要对原发病因进行重新评估。在进一步完善基因检测后,明确原发病为FMPP 的诊断。在临床诊疗如遇GnRHa 治疗效果欠佳的男性CPP 患者,应注意询问家族史,及时重新评估病因,调整治疗方案,尽早控制青春发育进程,避免成年身高矮小、行为问题以及肿瘤风险增加等不良后果。而本研究的病例2诊断CPP 的年龄小于4岁,小年龄段的中枢性性早熟,往往需高度怀疑继发于特殊病因,如生殖细胞瘤、先天性肾上腺皮质增生症以及LHCGR 基因突变导致的性早熟。
四、治疗
FMPP 的治疗以对抗其雄激素作用以及抑制甾体类性激素合成为主,治疗的目的是抑制过早及过快的第二性征发育,延缓骨龄进展,改善终身高,并减少因性早熟所致的社会或心理问题。目前用于治疗FMPP 的药物主要包括抗雄激素药物、P450 酶抑制剂以及芳香化酶抑制剂[38-40],但尚无统一的诊疗共识,临床上多采用联合用药方案。若患者继发CPP,可使用GnRHa 联合治疗,以期获得最大终身高获益。
本研究病例1 初治时仅使用GnRHa 抑制下丘脑-垂体-性腺轴功能,但治疗半年青春发育仍在进展,明确病因诊断后加用抗雄激素药物氟他胺及芳香化酶抑制剂来曲唑,抑制外周雄激素作用,并且阻断雄激素向雌激素转化,骨龄进展得到明显控制,生长速率逐步减慢,预测终身高较前有所改善。本研究中,病例2 明确病因诊断后开始治疗,初始方案为螺内酯+GnRHa 治疗。由于芳香化酶抑制剂对生殖系统以及骨代谢的长期影响仍未明确,家长起初拒绝使用芳香化酶抑制剂。而在初始方案治疗半年后,睾丸退缩不明显,睾酮水平仍高,骨龄出现明显提前,故加用第三代芳香化酶抑制剂阿那曲唑治疗,联合治疗半年后,患儿的睾酮水平明显下降,骨龄基本控制。
本研究的患者均为临床诊断CPP,进一步明确病因为LHCGR 基因突变所致性早熟继发CPP。对于男性性早熟患者,应注意评估其起病年龄、家族史以及治疗效果监测,必要时可完善基因检测,尽可能明确病因诊断,避免漏诊、误诊。
猜你喜欢
现代泌尿生殖肿瘤杂志(2022年1期)2022-11-21
中国实用医药(2022年18期)2022-09-20
中国临床医学影像杂志(2022年5期)2022-07-26
保健与生活(2022年10期)2022-05-06
科学24小时(2020年5期)2020-08-02
中国生殖健康(2019年7期)2019-01-06
心血管外科杂志(电子版)(2018年1期)2018-11-08
中国体育教练员(2017年3期)2018-01-19
中国体育教练员(2017年2期)2017-07-31
中国男科学杂志(2016年5期)2016-12-01
