
基于生物信息学的结直肠腺癌miRNA预后模型的建立
2022-02-18 12:52赵栋燕尹腾飞姚树坤
胃肠病学和肝病学杂志 2022年1期
赵栋燕, 尹腾飞, 姚树坤
1.中国医学科学院 北京协和医学院,北京 100029;2.中日友好医院消化内科;3.北京大学医学部
结直肠癌是全球最常见的恶性肿瘤之一,占每年全球新发癌症及癌症相关死亡人数的10%。近年来结直肠癌的经济负担在发展中国家逐渐增加,预计到2030年新病例的数目将超过220万,死亡人数将达到110万[1]。结直肠癌最常见的病理类型是腺癌,占所有类型的70%~90%,甚至更高[2]。尽管目前化疗、手术、靶向治疗、放疗等多种治疗方法在一定程度上提高结直肠癌患者的生存率并减少其复发,但晚期结直肠癌患者的5年生存率仍然很低[1]。近年来结直肠癌的发病机制和分子机制等相关研究取得了巨大进展,但依然无有效的方法来预测患者的生存情况,因此,亟待可靠的分子模型来预测患者的预后,并指导治疗。
miRNA是非编码RNA家族的重要成员之一,由18~25个核糖核苷酸组成,通过与靶mRNA的3’端非编码区或5’端非编码区互补结合,抑制翻译或者使靶mRNA降解或沉默。据了解,miRNA调控着60%的人类蛋白编码基因组的表达,参与包括癌症在内的多种生物和病理过程,在细胞增殖、迁移、侵袭和转移中具有重要作用[3]。已经证实,miRNA在每一种人类肿瘤组织中均显示出异常表达模式,包括结直肠癌。而特定的miRNA具有抑癌或促癌作用,这意味着miRNA是肿瘤形成的关键因素[4]。在人类肿瘤的发生发展中发挥着重要作用。随着基因测序技术的不断进步,大量的miRNA不断被发现。越来越多的证据显示,miRNA对结直肠癌的生长、增殖和转移至关重要。比如,miR-496作用于靶基因RASSF6,参与调控WNT通路,诱导上皮间充质转化,促进结直肠肿瘤转移[5]。miR-506促进靶基因NR4A1的降解,从而抑制结直肠肿瘤细胞的生长、增殖与迁移[6]。有研究证实单个或多个miRNA可以作为肿瘤的预后因素。生物信息学是分子生物学和信息技术的结合体,它通过收集和筛选基因组序列信息,对其进行分析和解释,最终获得生物系统的规律。因此,本研究目的在于依据生物信息学技术应用癌症基因组图谱(The Cancer Genome Atlas,TCGA)建立能够有效预测结直肠腺癌患者预后的miRNA风险模型。
1 资料与方法
1.1 资料获取通过TCGA数据库下载结直肠腺癌患者与正常对照组的miRNA表达数据、mRNA表达数据及相关的临床数据(https://portal.gdc.cancer.gov/repository)。TCGA数据库向公众免费开放,因此,本研究不需要额外的伦理证明。
1.2 差异mRNA与差异miRNA的筛选应用R语言的“edgeR”包对miRNA和mRNA的表达谱进行归一化处理,比较肿瘤组与正常组的miRNA和mRNA表达情况,并筛选出差异的mRNA与miRNA,筛选标准为校正P值(FDR)<0.05和|log2FC|>1。
1.3 预后模型的构建与验证根据数据分析的要求,无完整临床特征信息(包括性别、年龄、生存时间、生存状态)及生存时间<30 d(表明可能由其他疾病引起的死亡)将排除在本模型构建之外。应用R语言的“caret”包将具有完整生存信息和差异表达miRNA表达谱的样本按照50%的比例随机分为训练集和测试集,“caret”包中的“createDataPartition”函数能够快速实现数据的分割。利用χ2检验检测训练集、测试集与合并数据集之间的临床病理信息有无差异。随后,对训练集的miRNA进行单变量Cox回归分析,筛选预后相关的miRNA。对于P<0.05的miRNA进行多变量Cox回归分析,构建了预测风险的模型:risk score=∑βi×expmiRNAi,其中expmiRNA是miRNA的表达水平,β是对应miRNA的回归系数。根据风险值的中位数进行分组,将所有肿瘤患者分为高风险组和低风险组。采用Kaplan-Meier法及Log-rank检验绘制生存曲线,绘制受试者工作特征曲线(receiver operating characteristic,ROC)计算对5年生存期预测的曲线下面积(area under the curve,AUC)来评估miRNA模型的效能,同时我们也计算了肿瘤TNM分期的预测生存预后的效能。通过多因素Cox比例风险模型分析判断miRNA预后模型是否为结直肠腺癌的独立风险因素。
1.4 miRNA靶基因的预测目前公认的miRNA靶基因预测网站有3个,包括miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/),targetScan(http://www.targetscan.org)与miRDB(http://www.mirdb.org/)。首先,从这三个网站下载了miRNA预测数据库,并使用Perl语言预测各个miRNA的靶基因,我们把同时存在于两个及两个以上数据库中的基因定义为miRNA的靶基因。为了明确这些靶基因是否参与结直肠腺癌的发病机制,将靶基因与结直肠腺癌中差异表达基因取交集,同时应用cytoscape软件(3.80版本)描绘miRNA与交集基因之间的关系。应用R语言的“clusterProfiler”与“org.Hs.eg.db”包对交集基因进行KEGG与GO富集分析。当P<0.05时,则认为富集的基因集有统计学意义。
1.5 筛选核心靶基因与生存相关的靶基因String数据库(https://string-db.org/)是一个搜寻蛋白质之间相互作用的数据库。应用该网站明确靶基因之间的互作关系,设置置信度参数为0.004,并下载蛋白互作关系的相关文件。应用cytoscape软件的插件cytoHubba明确前10个核心基因,并绘制网络关系图。最后,应用Kaplan-Meier法及Log-rank检验筛选出生存相关的靶基因。
2 结果
2.1 筛选差异mRNA与miRNA我们对531个肿瘤样品与11个正常样品进行了miRNA差异表达分析,筛选出520个差异表达的miRNA,其中上调的有328个,下调的有192个。随后,我们对554个肿瘤样品与48个正常样品进行了mRNA差异表达分析,筛选出7 677个差异表达基因,其中上调的有4 321个,下调的有3 356个。
2.2 预后模型的构建将所有具有完整临床病理信息的结直肠腺癌患者(n=484)随机分为训练集(n=244)与测试集(n=240)。两组患者在年龄、性别、TNM分期、随访时间、随访结局方面比较,差异均无统计学意义(见表1)。单因素Cox回归分析发现,在训练集中,共有13个miRNA与患者的总生存期密切相关(P<0.05)。通过单因素和多因素Cox回归分析,最终从13个候选基因中挑选出来7个miRNA用于构建风险模型(见表2)。Kaplan-Meier曲线显示这7个miRNA均与患者的总生存期密切相关(P<0.05)(见图1)。风险评分计算如下:风险评分=hsa-miR-891a-5p的表达值×0.201+hsa-miR-664b-3p的表达值×0.564+hsa-miR-485-5p的表达值×0.386-hsa-miR-486-5p的表达值×0.298-hsa-miR-3615的表达值×0.277-hsa-miR-21-3p的表达值×0.394-hsa-miR-3677-3p的表达值×0.372。

表1 484例结直肠腺癌患者的临床病理特征
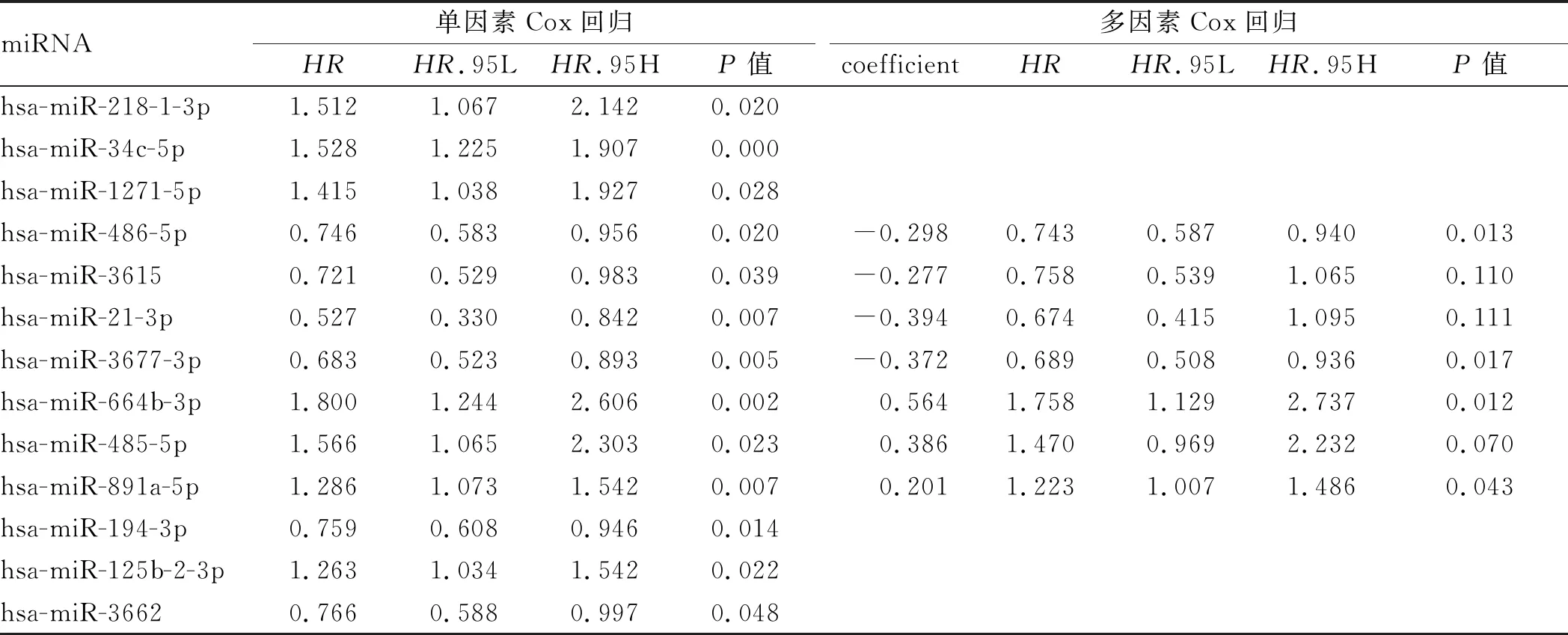
表2 差异miRNA的单因素与多因素Cox回归分析

图1 Kaplan-Meier曲线与Log-rank检验提示7个miRNA均与结直肠腺癌患者的总生存期显著相关
2.3 预后模型的验证在训练集中,根据miRNA风险模型计算了每一例结直肠腺癌患者的风险评分。以中位风险评分为截止点,将患者分为高风险组和低风险组,进行Kaplan-Meier分析,结果提示高风险组患者的总生存时间明显短于低风险组患者(P<0.001)。把风险公式应用于测试集与合并数据集之后,我们发现了类似具有显著性差异的结果(P<0.001)。根据患者的生存状态分布图,说明了测试集、训练集和合并数据集中低风险的患者生存时间长,预后佳(见图2)。另外,我们应用了ROC曲线对5年生存期进行了预测。如图2所示,在训练集中,miRNA预后模型对5年生存期的预测AUC为0.722;在测试集中,对5年生存期的预测AUC为0.747;在合并数据集中,对5年生存期的预测AUC为0.735,高于肿瘤TNM分期对5年生存期的预测AUC值(0.726)。
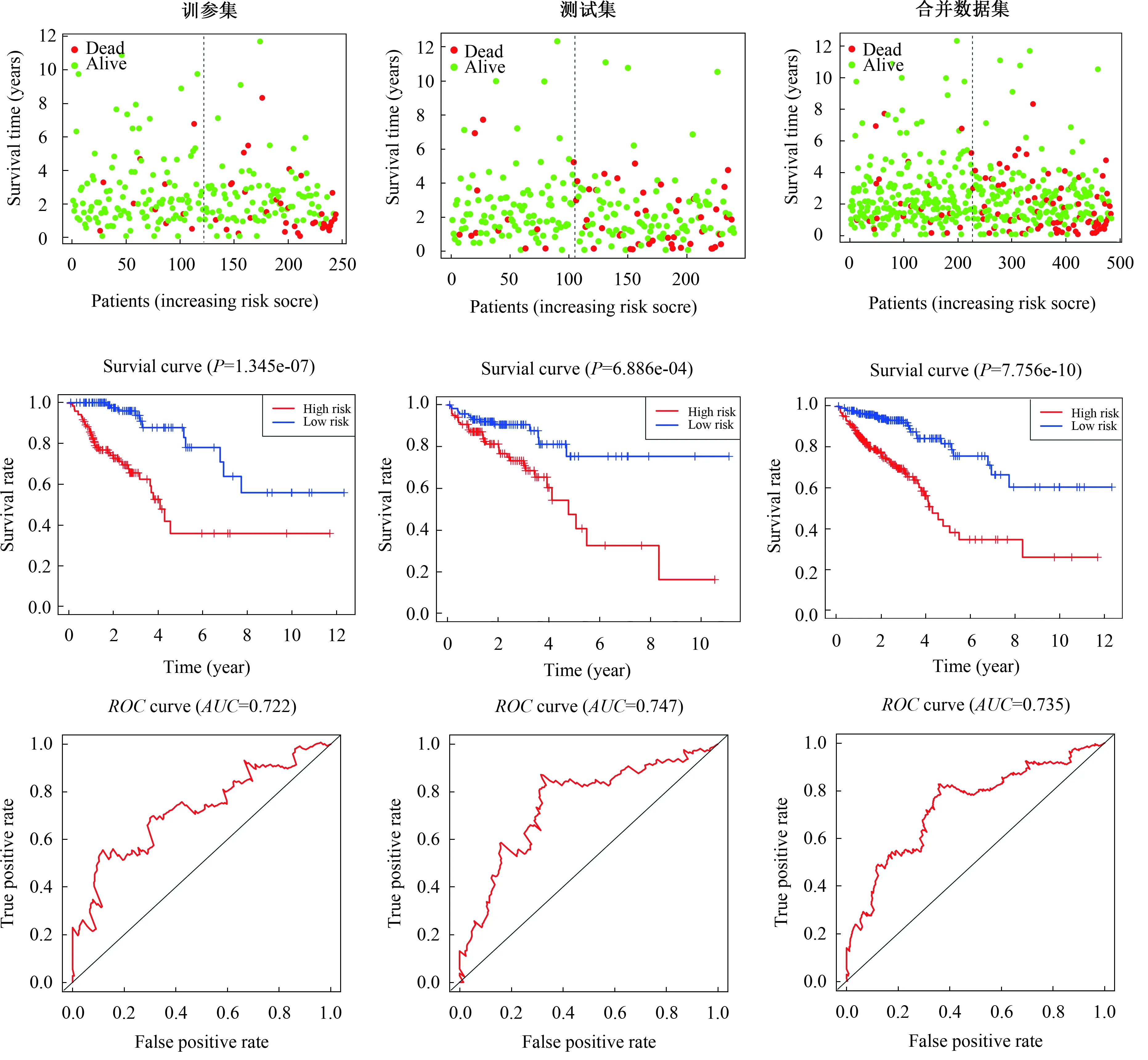
图2 训练集、测试集和合并数据集的Kaplan-Meier生存曲线、ROC曲线及生存状态分布图
为了明确miRNA风险模型是否为结直肠腺癌的独立预后风险因素,多因素Cox比例风险模型分析了miRNA模型与其他临床病理因素的相关性。在合并数据集中,我们发现miRNA模型是结直肠腺癌的独立风险因素,不受包括年龄、性别、TNM分期在内的临床病理参数的影响(HR=1.106,95%CI:1.033~1.185,P=0.004)(见图3)。

图3 miRNA预后风险模型的单因素Cox回归分析(A)与多因素Cox回归分析(B)
2.4 miRNA靶基因的预测通过3个靶基因数据库,我们预测了构建模型的7个miRNA所调控的靶基因。为了提供生物信息分析的准确性,对重叠的靶基因进行了鉴定。维恩图提示hsa-miR-891a-5p、hsa-miR-664b-3p、hsa-miR-485-5p、hsa-miR-486-5p、hsa-miR-3615、hsa-miR-21-3p和hsa-miR-3677-3p的靶基因数目分别是52个、49个、1 281个、344个、75个、631个、38个。接着,将所有的靶基因与7 677个差异表达基因进行交集,得到了277个交集基因。其中,112个基因在结直肠腺癌中表达下调,165个基因表达上调。图4展示了用于构建模型的7个miRNA与靶基因之间的网络关系。
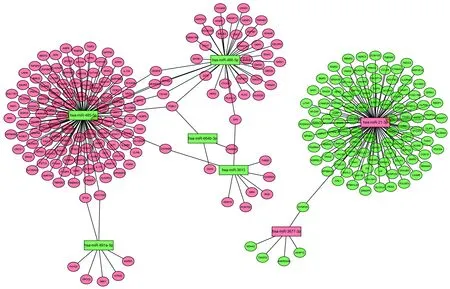
注:长方形代表miRNA,圆形代表mRNA;红色代表表达上调,绿色代表表达下调。
2.5 肿瘤相关靶基因的功能富集分析GO注释提示共有583个通路与肿瘤相关靶基因密切相关。GO结果中主要包括生物学过程(BP)、细胞组分(CC)和分子功能(MF)三个部分,每个部分前30个通路在图5中进行了展示。BP分析提示,靶基因与细胞生长、成骨作用、发展细胞生长等密切相关(见图5A);CC分析提示,靶基因富集在轴突生长锥、顶端质膜及含胶原的细胞外基质(见图5B);MF分析提示靶基因调控受体配体活性、信号受体激活因子活性、轴突导向因子结合、细胞因子活性等(见图5C)。KEGG分析提示共有18个通路与靶基因密切相关,比如Hippo信号通路、cGMP-PKG信号通路、轴突导向、长程增强效应及细胞因子受体相关作用等(见图5D)。
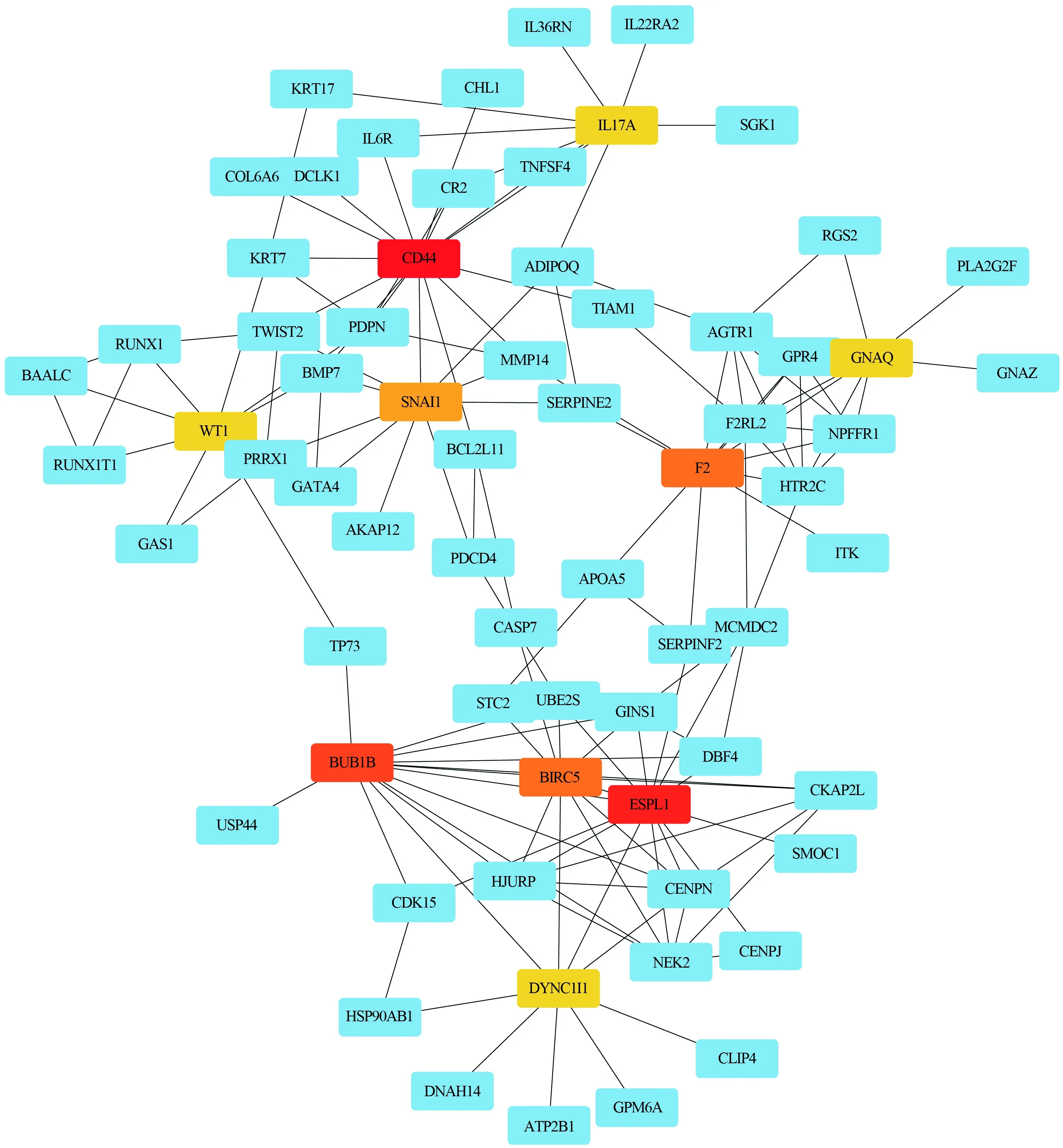
2.6 生存相关的靶基因Kaplan-Meier曲线提示277个肿瘤相关的靶基因中,其中有9个靶基因(DNA2、F2、F2RL2、HERC3、LITAF、SEMA6A、SEMA6D、XPNPEP3、ZNF831)与患者的生存预后呈正相关,有14个靶基因(C2orf48、FSTL3、LDLRAD3、MAB21L1、NPR3、ONECUT2、PLXNA3、RGS2、RNF150、SNAI1、SP5、SPIN3、STC2、TTYH3)与患者的生存预后呈负相关(见图6)。接着,将277个肿瘤相关的靶基因输入到String数据库中,描绘基因间的互作网络,并应用cytoscape软件筛选出10个核心基因(CD44、ESPL1、BUB1B、BIRC5、F2、SNAI1、WT1、DYNC1I1、GNAQ、IL17A)(见图7)。
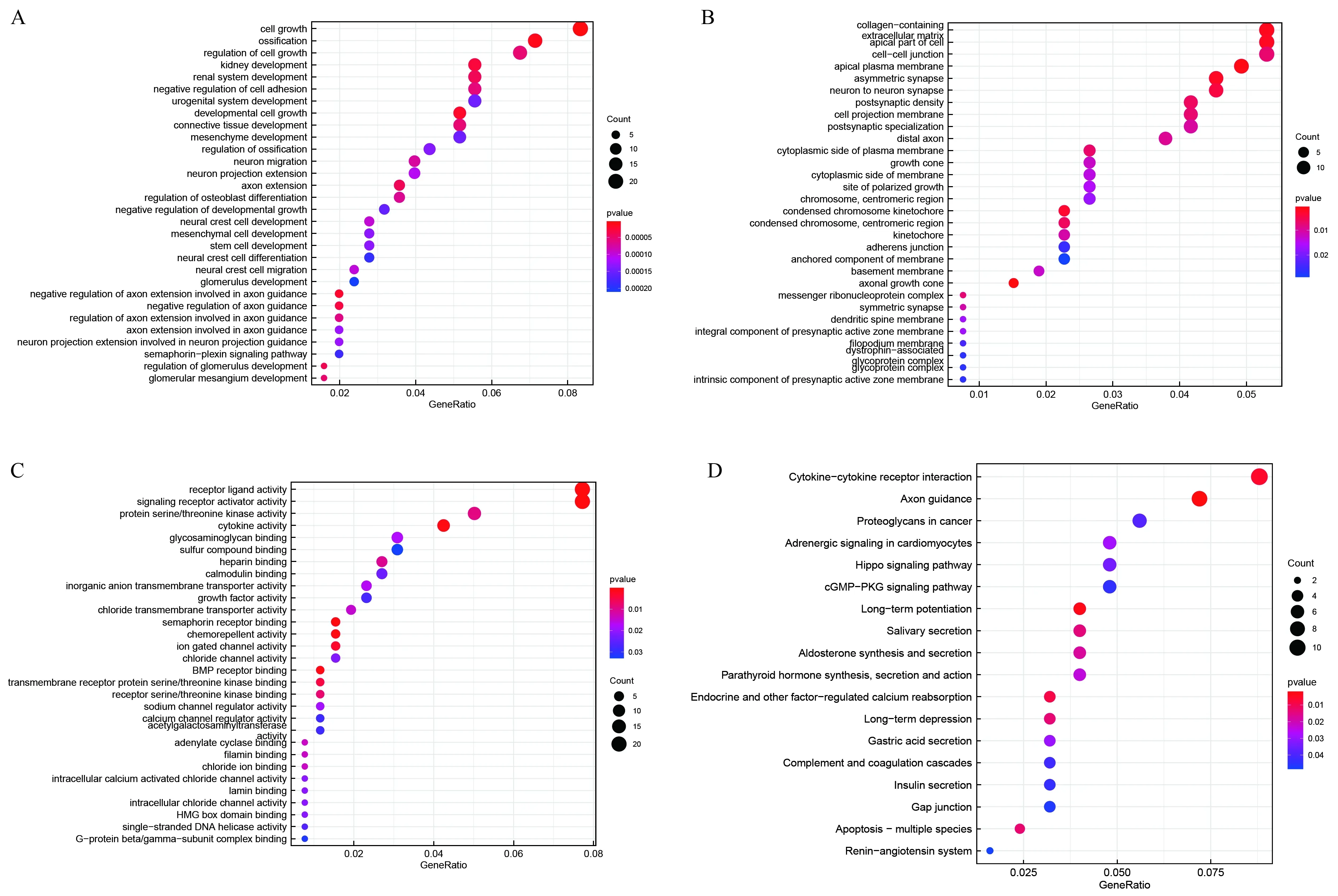
注:A:BP分析;B:CC分析;C:MF分析;D:KEGG分析。
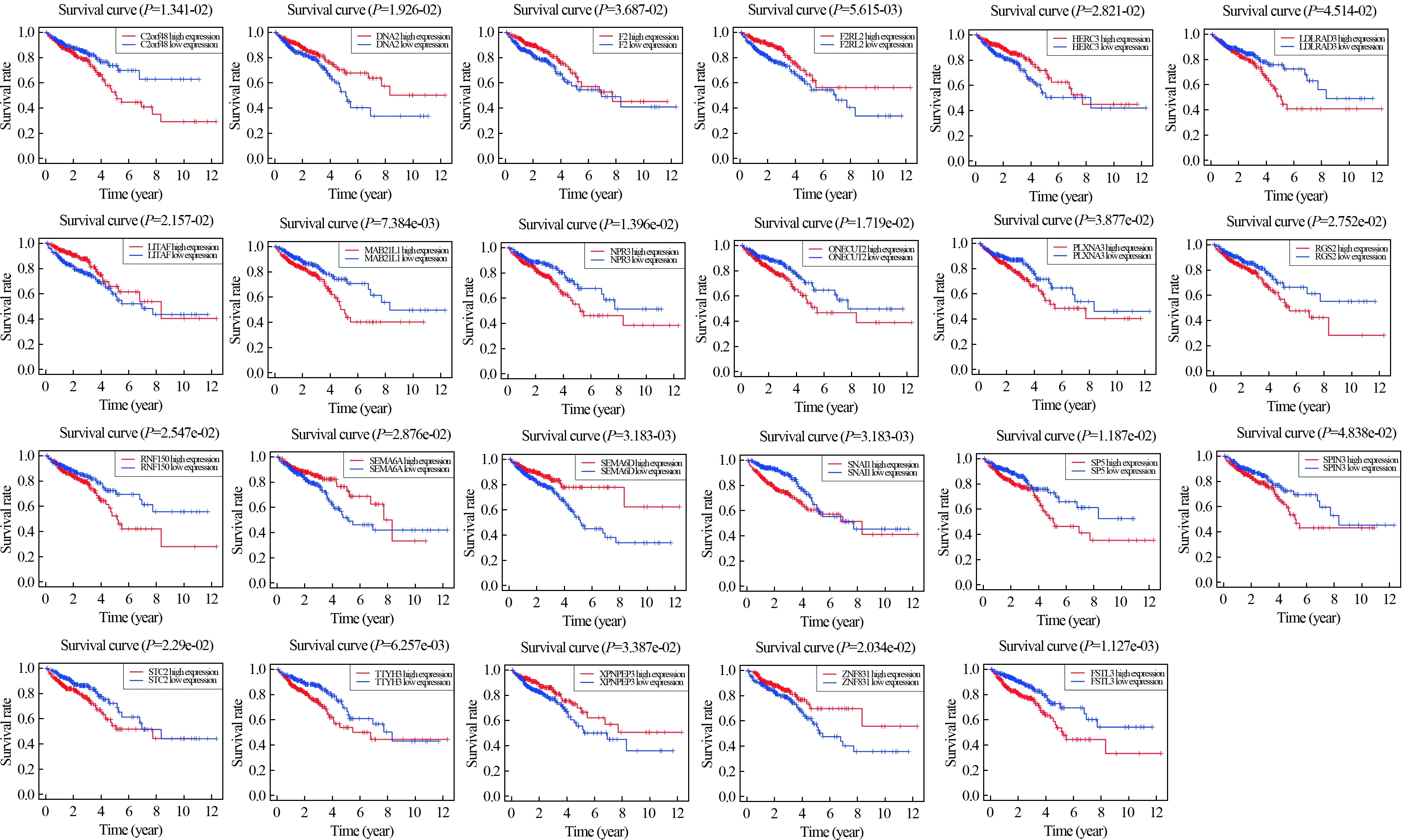
图6 Kaplan-Meier曲线与Log-rank检验提示23个靶基因与结直肠腺癌患者的总生存期显著相关
注:模块颜色越深,关联系数越高。
3 讨论
结直肠癌是一种高度恶性肿瘤,特别容易发生肝、肺转移,严重影响患者的生存预后。考虑到结直肠癌具有较高的肿瘤异质性,基于常规分期比如TNM分期无法准确预测疾病的转移、进展和临床结局。尽管肿瘤患者可能处于同一TNM期,但其临床结局可能存在较大差异。因此,寻找一种准确的预后生物标志物对了解结直肠癌的发病机制、预测临床结果和个体化治疗非常重要。因此,寻找一种高特异性和灵敏性的预后标志物对患者至关重要。由于miRNA在人体中广泛分布并能够稳定存在,miRNA被认为是一种的新型生物学标志物。越来越多的研究表明,miRNA参与调控结直肠癌的血管生长、上皮间充质转化、过度增殖、凋亡缺失、干细胞稳态等多个病理生理机制,是结直肠癌的治疗靶点和预后指标[7]。因此,基于TCGA数据库,我们建立了由7个miRNA组成的能够预测结直肠腺癌患者预后的风险模型。
首先,我们从TCGA数据库下载了结直肠腺癌患者的成熟miRNA表达谱、转录组表达谱及相应的临床资料。通过差异表达分析,获得了520个差异miRNA和7 677个差异mRNA。所有的患者被随机分配为训练集和测试集,在训练集中通过单因素、多因素Cox分析建立了由7个miRNA(hsa-miR-891a-5p、hsa-miR-664b-3p、hsa-miR-485-5p、hsa-miR-486-5p、hsa-miR-3615、hsa-miR-21-3p与hsa-miR-3677-3p)组成的预后模型。同时,我们在训练集、测试集和合并数据集中验证了该miRNA模型的预后效能。Kaplan-Meier曲线提示,在三个数据集中均呈现出高风险组患者的生存率显著低于低风险组的趋势。ROC曲线提示模型预测5年生存率的AUC均>0.7,并且高于肿瘤TNM分期的AUC值,表明该模型具有较好的效能。此外,多因素Cox比例风险模型分析提示miRNA模型是结直肠腺癌的独立风险因素,不受其他临床病理因素如年龄、性别、TNM分期等的影响。
既往研究[8-17]表明,用于构建模型的miRNA均参与调控肿瘤的生长、发育、侵袭与转移等功能。比如,hsa-miR-891a-5p是激素受体阳性乳腺癌与非小细胞肺癌的新型生物学标志物,能够调控肿瘤的生物学功能[8-9]。一个小样本研究表示hsa-miR-664b-3p的升高常伴随着胰腺神经内分泌肿瘤远处转移的出现,可以作为预测胰腺内分泌肿瘤转移与否的一种潜在的生物标志物[10]。hsa-miR-485-5p作为一种抑癌基因抑制多种肿瘤细胞的生长与侵袭,并能增加化疗药物的敏感性,比如乳腺癌[11]、卵巢癌[12]、食管癌[13]。与既往研究相反的是,本研究中hsa-miR-485-5p的表达水平与患者的生存时间呈负相关,提示hsa-miR-485-5p在结直肠腺癌中能够促进肿瘤生长。有研究表明,hsa-miR-486-5p作用于靶基因PIK3R1抑制结直肠癌细胞的迁移与侵袭,可以作为结直肠癌的治疗靶点[14]。一项基于生物信息学的研究提出hsa-miR-3615与肝癌患者的总生存时间呈负相关,与TNM高分期、血清AFP与Ki67水平呈正相关[15]。而在本研究中,hsa-miR-3615与结直肠腺癌患者的总生存时间呈正相关,提示其在不同肿瘤中可能发挥着不同的作用。一项基础研究提出miR-21-3p可能通过靶向NAV3基因在卵巢肿瘤中诱导顺铂耐药[16]。发表在2020年的一项研究提示缺氧诱导的miR-3677-3p能够通过抑制SIRT5促进肝细胞癌细胞的增殖、迁移和侵袭[17]。然而,这些miRNAs在结直肠腺癌中到底发挥什么样的作用,未来还需要大量的临床与基础研究来进行探索。
为了进一步了解用于构建模型的miRNA在结直肠腺癌中的作用机制,我们首先对miRNA的靶基因进行了预测,随后将靶基因与差异的mRNA取交集,对交集基因进行了功能富集分析。GO结果提示交集基因主要与细胞生长、成骨作用、发展细胞生长、轴突生长锥、顶端质膜、含胶原的细胞外基质、信号受体激活因子活性、轴突导向因子结合及细胞因子活性等有关。KEGG结果提示交集基因参与调控Hippo信号通路与cGMP-PKG信号通路。大量研究表明这两条通路参与调控人类肿瘤的生物学功能[18-20]。以上结果提示,我们用来构建预后模型的miRNA参与调控肿瘤相关的信号通路,为预后模型的临床应用提供了生物学的证据。
为了明确miRNA预后模型调控结直肠腺癌的关键机制,我们用cytoscape软件的插件cytoHubba筛选出10个核心基因,包括CD44、ESPL1、BUB1B、BIRC5、F2、SNAI1、WT1、DYNC1I1、GNAQ与IL17A。与此同时,我们应用Kaplan-Meier法绘制了所有靶基因的生存曲线,结果提示共有23个靶基因与结直肠腺癌患者的总生存期显著相关。值得注意的是,靶基因F2不仅是蛋白互作网络的核心基因之一,也与患者的预后生存显著相关。这些结果提示本研究中用于构建模型的miRNA可能通过调控靶基因F2来影响肿瘤患者的预后生存。F2基因编码凝血酶原蛋白,又称为凝血因子Ⅱ。凝血因子Ⅱ是一种多结构域糖蛋白,对生命至关重要,是抗凝治疗的关键靶点[21]。在癌症患者中,高凝状态是一个常见的现象。它不仅与静脉血栓栓塞的风险增加有关,也与肿瘤增殖和进展有关[22]。然而,目前无研究证实F2参与调控结直肠癌的发生发展。未来需要进一步的试验去探索模型相关的miRNA与靶基因在结直肠癌中的功能。
本研究存在一些不足之处。我们的研究仅仅纳入了TCGA这一个数据库,未用外部数据库比如GEO数据库(Gene Expression Omnibus database)或其他大样本的临床队列进行验证。另外,我们应用三个靶基因数据库预测了miRNA的靶基因,并未进行基础实验在细胞和分子水平进一步的验证。
基于生物信息分析,我们成功地构建了一个能够有效预测结直肠腺癌患者预后的miRNA模型,并证实该模型优于常规肿瘤分期。更重要的一点是,该模型不仅与患者的总生存期显著相关,而且是结直肠腺癌的独立风险因素。另外,我们通过对构建模型的miRNA靶基因进行预测,推断了miRNA的潜在生物学功能具有调控结直肠腺癌的发生、发展、增殖及转移的潜力,寻找了该模型调控的关键基因,为结直肠腺癌的病理生理机制研究提供了一个新的方向。
猜你喜欢
延安大学学报(医学科学版)(2022年2期)2022-11-23
临床军医杂志(2022年7期)2022-08-06
基层中医药(2022年1期)2022-07-22
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
现代临床医学(2022年1期)2022-02-12
家庭医药·快乐养生(2021年8期)2021-08-30
中国现代医生(2016年31期)2017-03-02
中国现代医生(2016年32期)2017-03-02
为了孩子(孕0~3岁)(2001年3期)2001-06-13
