
壳聚糖-海藻酸钠水凝胶的制备及其在制革含Cr(III)废水治理-复鞣填充工段的潜在应用
2022-02-14 05:40官小玉张炳原常金明贺梦妍陈咏梅
陕西科技大学学报 2022年1期
官小玉, 张炳原, 常金明, 贺梦妍, 安 盟, 陈咏梅
(1.陕西科技大学 轻工科学与工程学院, 陕西 西安 710021; 2.陕西科技大学 中国轻工业皮革清洁生产重点实验室, 陕西 西安 710021; 3.西华师范大学 化学合成与污染控制四川省重点实验室, 四川 南充 637009; 4.陕西科技大学 机电工程学院, 陕西 西安 710021)
0 引言
制革鞣制,是鞣剂分子向皮内渗透并与生皮胶原分子活性基团结合而发生性质改变的过程.长期以来,由于铬鞣能赋予成革众多优良性能,如耐湿热稳定性高,耐水洗能力强,柔软、丰满、有弹性等,铬鞣剂备受青睐[1-3].然而,皮胶原对传统铬鞣剂的吸收能力有限,约有30%~40%的铬(Cr(III))被排放于废水中,对环境造成严重污染且对铬资源造成极度浪费.在众多Cr(III)治理方法中,吸附法凭借其操作简便、治理效率高、可再生等优点而被广泛地研究与应用[4-7].传统吸附剂,如天然矿物质、无机纳米材料、农业废弃物、微生物生物质类等[6,8-13],由于比表面积大、亲和性强、成本低等优势在Cr(III)吸附领域占有重要地位.然而,这类传统吸附剂在Cr(III)治理结束后,难以得到进一步的资源化再利用,与环保可持续发展理念背道而驰.
另一方面,针对吸附后的Cr(III)或其他重金属离子,通常采用酸(如硝酸[14,15]、盐酸[16,17])、碱(如氢氧化钠[18,19])或螯合剂(如EDTA[20])等化学物质,通过改变吸附剂表面的电荷或存在竞争关系从而使吸附了的金属离子从吸附剂表面脱落,然后从溶液中回收金属离子,进一步加入其他络合配体,生成含铬(或其他金属离子)配合物.例如,在回收后的Cr(III)溶液中加入硫酸与氢氧化钠可制备成碱式硫酸铬,进一步用于制革鞣制领域.然而,传统的化学脱附-重新整合-再利用方式存在操作复杂且耗时等不足.
水凝胶是物理或化学交联而成的具有三维网络结构的高分子材料,在水中能够溶胀,吸水量可达凝胶干重的几十倍甚至几千倍,是一种特殊的半固体材料[21-23].通过选择适当的亲水主链,可赋予水凝胶对特定污染物良好的吸附能力,且易分离,可重复使用,广泛应用于污水处理领域[24-27].
水凝胶一般包括天然生物质水凝胶(如壳聚糖类、海藻酸钠类、植物纤维基类、明胶类等)以及合成水凝胶(如聚丙烯酸类、聚丙烯酰胺类、聚乙烯醇类等).水凝胶吸附材料的吸附能力与其溶胀率息息相关,虽然合成类水凝胶具有很强的吸水溶胀性能,但是生物可降解性差,生物毒性大,在一定程度上限制其在污水处理领域的应用.
壳聚糖是由甲壳素脱乙酰化得到的一种天然碱性多糖,自然界储量极为丰富.海藻酸钠是从褐藻类的海带或马尾藻中提取碘和甘露醇之后的副产物,其分子由β-D-甘露糖醛酸(β-D-mannuronic,M)和α-L-古洛糖醛酸(α-L-guluronic,G)按(1→4)键连接而成,也是一种天然多糖.近年来,由于壳聚糖与海藻酸钠的生物相容性好、潜在毒性低、微生物降解性好,产品废弃后对环境的影响较小等优势,在生物医学、组织工程、仿生工程、水处理等领域得到广泛的应用[28-31].将壳聚糖与海藻酸钠天然生物质类水凝胶作为金属离子吸附剂具有三大优势:首先,水凝胶结构中带有大量的活性功能基团,如羧基、羟基、氨基等,他们能通过配位或静电作用实现金属离子的快速吸附;其次,水凝胶的空间网状结构赋予其大比表面积、多结合位点等特性,可大大提高吸附效能;此外,壳聚糖与海藻酸钠水凝胶的可降解性能可杜绝吸附剂的二次污染问题.
综上分析,针对传统吸附法治理含铬废水存在吸附剂难降解、循环再利用过程复杂且耗时等不足,本论文提出基于天然生物质水凝胶的含铬(Cr(III))制革废水治理及资源化利用方法.本论文以天然生物质水凝胶海藻酸钠(SA)作为聚阴离子组分,壳聚糖(CS)作为聚阳离子组分,戊二醛(GA)为交联剂,采用内凝胶法制备了海藻酸钠-壳聚糖(CS-SA)水凝胶.通过对比研究海藻酸钠、壳聚糖组分比例,戊二醛用量以及干燥方式,来优化制备CS-SA水凝胶,使其在具有较稳定结构的基础上兼具疏松的内部结构.一方面,疏松的内部结构赋予CS-SA水凝胶大比表面积、多结合位点等特性,可潜在提高Cr(III)的吸附效能;另一方面,疏松的空间网状结构利于CS-SA-Cr(III)金属凝胶的降解,即更利于凝胶向CS/SA/Cr(III)溶胶转变.将CS/SA/Cr(III)溶胶应用于制革复鞣填充,基于CS、SA、Cr(III)以及GA的鞣制效能,可潜在提高成革的耐湿热稳定性;基于CS、SA、GA的填充性能,在理论上,成革的粒面会更加紧实平细,革身更柔软丰满.因此,本论文优化制备的CS-SA天然生物质水凝胶可潜在实现集Cr(III)废水治理-皮革复鞣填充一体化,从而实现吸附剂-吸附质最大资源化利用.
1 实验部分
1.1 材料与仪器
1.1.1 主要材料
壳聚糖(CS,200~400 mpa·s,中国上海阿拉丁工业公司);海藻酸钠(SA,200±20 mpa·s,中国上海阿拉丁工业公司);戊二醛(GA,25 g/L,天津市科密欧化学试剂有限公司);冰醋酸(250 g/L,天津市富宇精细化工有限公司).
1.1.2 主要仪器
电子天平(FA2004,上海舜宇恒平科学仪器有限公司);DF-1集热式恒温磁力搅拌器(XMTD-702,金坛市江南仪器厂);电热鼓风干燥箱(WGL-1258,天津市泰斯特仪器有限公司);冷冻干燥机(Xinyi-10N,宁波新艺超声设备有限公司);扫描电子显微镜(TESCAN,捷克TESCAN);BrukerV70型傅里叶变换红外光谱仪(BrukerV70,德国布鲁克公司).
1.2 海藻酸钠-壳聚糖水凝胶(CS-SA)的制备
1.1.2 主要仪器
以戊二醛(GA)为交联剂,CS作为聚阳离子组分,SA作为聚阴离子组分,制备CS-SA水凝胶.然而,常用制备凝胶的方法即将冰乙酸直接加入CS/SA体系,这样聚阴离子和聚阳离子的瞬时作用通常会导致不均匀的沉淀,使得制备理想形状的水凝胶非常困难.为解决这个问题,本实验采取缓释的方法将一定质量的SA溶于去离子水中,磁力搅拌机室温下搅拌50 min至完全溶解,同时将适量CS溶于去离子水中,磁力搅拌机室温搅拌50 min后,将两种溶液混合,加入适量GA,搅拌10 min 形成浆状溶液,放入模具并暴露在酸性室温环境中反应24 h.当酸性蒸汽扩散到混合物的表面和内部时,CS缓慢质子化,避免形成不均匀的凝胶.之后浸入去离子水中,除去未反应的GA小分子,干燥得到干态水凝胶.具体的反应步骤与条件控制如下,反应示意图如图1所示:
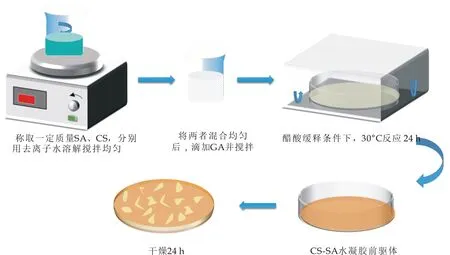
图1 CS-SA水凝胶制备路线图
(1)称取0.2~0.6 g CS,0.2~0.6 g SA,分别在一定量的去离子水中搅拌50 min直至体系分散均匀;
(2)将搅拌均匀的CS、SA溶液倒入烧杯中搅拌10 min混合均匀;
(3)量取0~0.5 mL GA加入CS/SA溶液中,搅拌5 min混合均匀;
(4)将CS/SA/GA溶液倒入圆形培养皿;量取25 mL的冰醋酸加入方形培养皿,并将圆形培养皿放入,调整水浴锅温度30 ℃,反应24 h得到CS-SA水凝胶前驱体;
(5)将得到的水凝胶前驱体用去离子水冲洗除去未反应的GA小分子,干燥24 h得到干态CS-SA天然生物质水凝胶.
1.3 CS-SA制备条件优化
在制备CS-SA水凝胶的合成条件基础上分别探究分析以下条件对水凝胶内在结构性能的影响.
(1)SA与CS(g)用量的优化
实验方法同上,控制其他条件不变(GA用量0.3 mL),调节CS与SA用量,具体优化方案如表1所示.
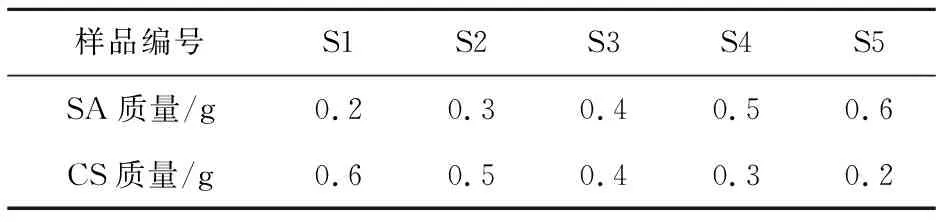
表1 SA与CS组分用量
(2)GA(mL)用量的优化
实验方法同上,控制其他条件不变(SA 0.6 g,CS 0.2 g),调节GA的用量,具体优化方案如表2所示.

表2 GA用量
(3)干燥方式的优化
冷冻干燥,恒温干燥箱干燥.
1.4 CS-SA表征
(1)采用红外光谱(FTIR)表征
室温下使用傅立叶变换红外光谱仪在衰减全反射模式下(ATR)对冻干的水凝胶样品进行测试.粉末样品表征:样品与预先在120 ℃干燥4 h的溴化钾1∶4在研钵中研磨混合均匀,并压制成均匀透光的压片进行红外表征.膜样品表征:将已干燥的样品截取一小块直接使用红外光谱仪进行测量.
(2)采用扫描电镜(SEM)观察
对冻干的海藻酸钠-壳聚糖水凝胶样品进行液氮脆断,并对断面进行喷金处理,之后用扫描电镜进行观察.
2 结果与讨论
2.1 CS-SA天然生物质水凝胶合成条件分析
2.1.1 反应方法
如图2所示,本论文合成的CS-SA水凝胶成胶机理包括两方面:首先是聚阳离子组分的CS与聚阴离子组分的SA通过静电相互作用聚集;当加入交联剂戊二醛(GA)以后,CS结构中氨基与GA结构中的醛基发生交联反应,生成席夫碱.值得注意的是,戊二醛在溶液中可能发生水解反应,进一步增长链段,因此通过GA相连的两条CS链之间距离增大.当然,GA也可能连接同一条CS链上的两个氨基,从而破坏壳聚糖大分子链的规整性.
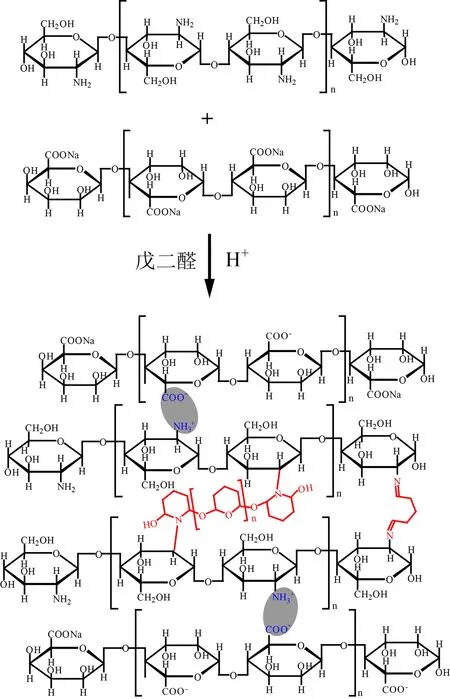
图2 CS-SA水凝胶合成反应方程式
常用制备CS-SA凝胶的方法,又称外凝胶法,即将冰乙酸直接加入SA/CS体系,这样聚阴离子和聚阳离子的瞬时作用通常会导致不均匀的沉淀(如图3(a)所示),使得制备理想形状的水凝胶非常困难.为了解决这个问题,本实验采取酸性缓释内凝胶法,将SA/CS浆状溶液暴露在酸性条件下,当酸性蒸汽扩散到混合物的表面和内部时,缓慢成胶,避免了不均匀沉淀物的形成(如图3(b)所示).
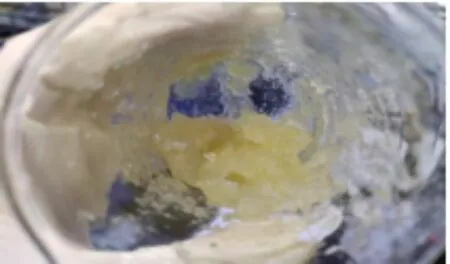
(a)外凝胶法制备CS-SA水凝胶
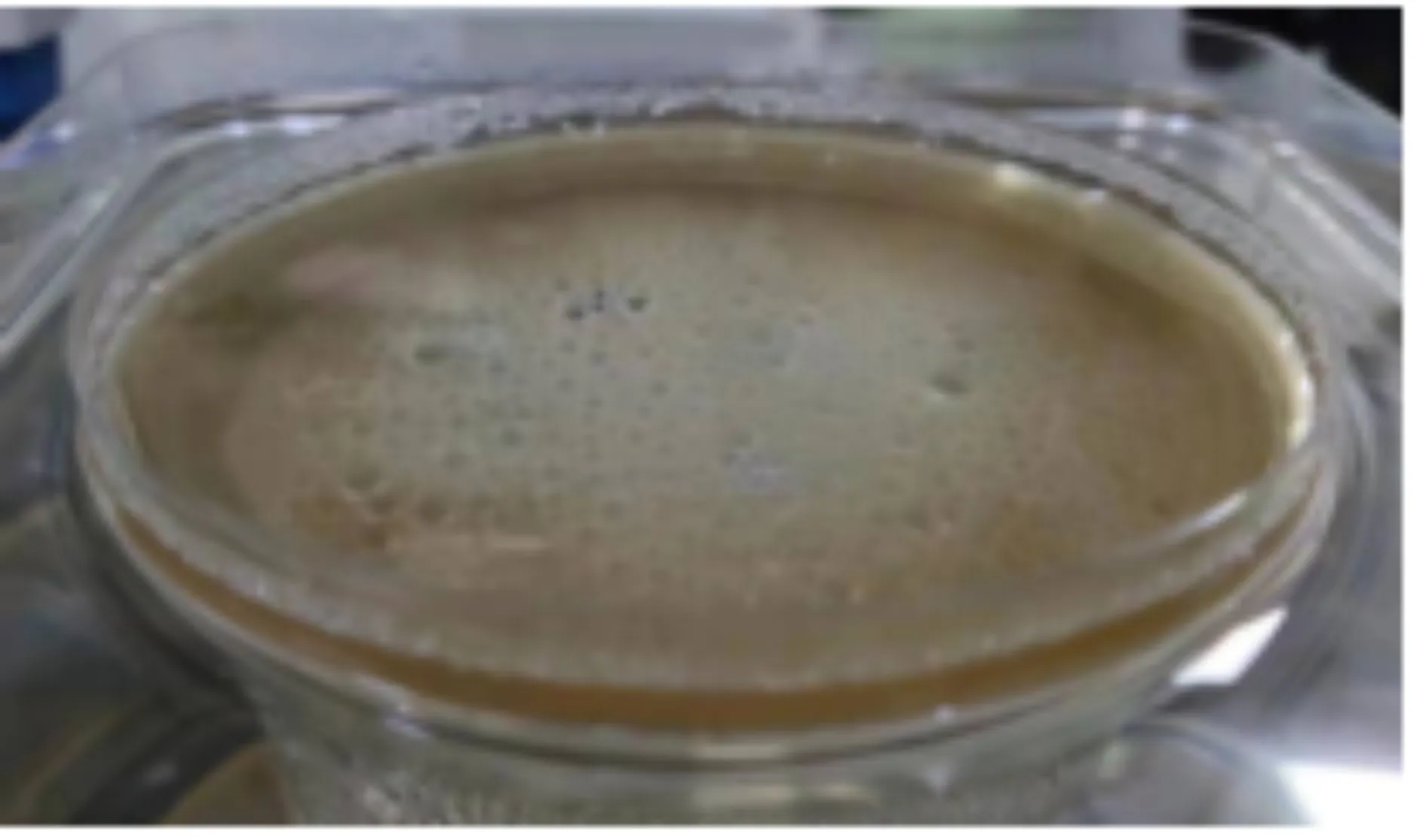
(b)内凝胶法制备CS-SA水凝胶图3 不同反应方法制备的CS-SA水凝胶外形图
2.1.2 海藻酸钠(SA)与壳聚糖(CS)用量
不同质量比的CS、SA对CS-SA天然生物质水凝胶能否成胶以及成胶后的性能有一定的影响.如图4所示,随着SA∶CS质量比增加,一方面,SA、CS体系更易成胶;另一方面,SA用量提高可增加功能基团-COO-的含量.基于-COO-与Cr3+间的配位吸附机理,-COO-含量提高可潜在提高CS-SA水凝胶对溶液中Cr(III)的吸附效能.然而,实验发现随着SA∶CS质量比增加,水凝胶成品在溶液中越稳定,稳定的CS-SA不利于后期的降解,尤其是通过简单的机械作用.综合考虑到CS-SA水凝胶既对溶液中Cr(III)具有较好的吸附效能又能在溶液中稳定一段时间且利于后期降解并用于制革复鞣填充,本论文最终选择SA与CS的质量分别为0.6 g与0.2 g,即SA与CS用量比为3/1.

图4 SA、CS质量比对成胶的影响
2.1.3 戊二醛(GA)用量
通过探究实验发现,水凝胶表面的形貌结构会随着交联剂GA的用量呈现明显的差异.如图5所示,交联剂GA用量为0 mL时水凝胶表面光滑,结构均匀而细密,而加入交联剂后,水凝胶表面结构疏松,网络结构清晰可见并且随着交联剂用量的增加,水凝胶的网络孔隙逐渐增大.造成水凝胶孔隙增大的主要原因是:戊二醛加入可使水凝胶内部形成互穿网络的结构,随着交联剂用量增加,交联度逐渐增大,壳聚糖大分子链的规整性受到破坏,导致整个结构变得疏松.疏松的孔隙结构能够赋予水凝胶更大的比表面积,为后期Cr(III)的吸附提供充足的结合位点,同时在吸附Cr(III)后,有利于CS-SA-Cr(III)金属凝胶降解成CS/SA/Cr(III)溶胶.然而,当交联剂用量超过0.3 mL时,由于壳聚糖大分子链之间的交联程度过高,使其规整性受到较大破坏,从而导致CS-SA在溶液中的稳定性大大下降,即在吸附Cr(III)过程中,CS-SA难以维持稳定.综合考虑到CS-SA的吸附效能、CS-SA-Cr(III)后期降解以及使用成本,本论文最终选择交联剂的用量为0.3 mL.

图5 GA用量对CS-SA水凝胶形貌的影响
2.1.4 干燥方式影响
在本论文中,分别采取常温烘箱干燥以及冷冻干燥的方式来考察干燥方式对CS-SA水凝胶的结构及性能影响.如图6所示,常温干燥后的水凝胶结构紧密,而冷冻干燥后的水凝胶结构蓬松,其主要原因是:常温干燥时,链段间的距离会随着水分的蒸发而逐渐塌陷并靠近,整个体系会随着水分的离开而变的更加致密(如图6(a)所示);而冷冻干燥时,水凝胶网络结构中的水在低温下立刻升华并会保持水凝胶原有的物理结构,因此结构会更加松散(如图6(b)所示).松散的内部结构可大大提高凝胶的比表面积及孔隙率,可潜在为后期Cr(III)的吸附提供充足的结合位点.另一方面通过水凝胶在水中的溶胀实验发现,相比于常温干燥,冷冻干燥后的水凝胶放入水溶液中更容易溶胀直至溶解,该现象表明冷冻干燥的方式在一定程度上可以促进水凝胶在溶液中的降解,为后续CS-SA-Cr(III)水凝胶的降解及复鞣填充提供便利.因此,本论文最终选择水凝胶的干燥方式为冷冻干燥.

(a)常温干燥 (b)冷冻干燥图6 干燥方式对CS-SA水凝胶结构性能的影响
2.2 CS-SA干态水凝胶表征
2.2.1 FTIR表征
本论文对SA、CS、CS-SA(含交联剂GA)以及CS-SA(不含交联剂GA)产品进行红外检测,其结果如图7所示.首先,CS在1 649 cm-1和1 591 cm-1处显示出两个吸收峰,分别归属于伯胺中C=O的拉伸振动和N-H的弯曲振动;SA在1 594 cm-1和1 406 cm-1处的吸收峰值可分别归属于羧基的不对称和对称拉伸振动.此外,相比于CS与SA,CS-SA(不含GA)的红外谱图显示1 649 cm-1和1 591 cm-1处的吸收峰值消失,可能是因为-NH2被质子化成-NH3+.另一方面,由于酸化过程中部分质子化的羧基,在CS-SA(不含GA)形成后,1 714 cm-1处出现一个新峰.以上结果证实了壳聚糖和海藻酸钠之间存在静电相互作用.
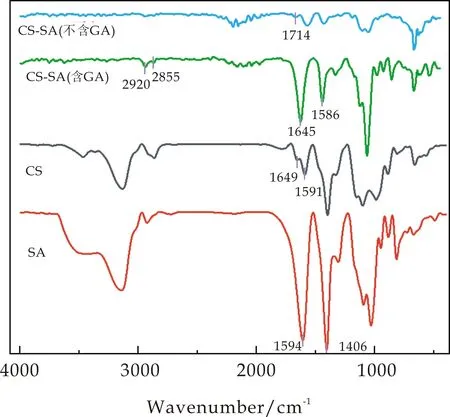
图7 CS-SA水凝胶的傅里叶变换红外光谱图
在合成CS-SA过程中,当加入交联剂GA后,CS-SA(含GA)在1 645 cm-1和1 586 cm-1处出现了亚胺键(C=N)的强伸缩振动吸收峰,为席夫碱的特征吸收峰,表明-NH2和HC=O的结合生成HC=N.此外,在2 920 cm-1和2 855 cm-1处的C-H振动峰明显,进一步表明CS与GA形成席夫碱交联网络.
综上,红外光谱结果表明在醋酸缓释的条件下,SA与CS间形成静电相互作用的物理交联,CS与GA之间形成了席夫碱交联网络,因此,最终成功地合成了以GA为交联剂的CS-SA天然生物质水凝胶.
2.2.2 SEM表征
对冷冻干燥后的海藻酸钠-壳聚糖水凝胶样品进行液氮脆断,并对断面进行喷金处理,然后用扫描电镜观察其微观结构.图8显示了不同GA(0 mL、0.3 mL 以及0.5 mL)含量的干态CS-SA水凝胶在500μm下的扫描电镜图像.从图8可以明显地看出,CS-SA水凝胶内部结构呈现多孔状.一方面,多孔结构有利于溶胀,进而促进重金属离子扩散到水凝胶内部与体系中的功能基团结合,实现对金属离子的吸附作用.另一方面,这些孔隙结构可增加CS-SA的比表面积及活性结合位点,进一步利于水凝胶对重金属离子的吸附.与此同时,对比图8(a)与图8(c),可以看出加入交联剂GA,即凝胶内部的交联程度提高,可使水凝胶网络结构松散,体系更加蓬松,该现象与图5结果吻合,进一步证明GA的引入,可促进CS-SA结构变得蓬松,既为Cr(III)的吸附提供更多的结合位点又利于后期体系的降解.

(a)CS-SA(GA 0 mL)

(b)CS-SA(GA 0.3 mL)

(C)CS-SA(GA 0.5 mL)图8 CS-SA水凝胶的扫描电镜图像
3 结论
在本研究中,以壳聚糖(CS)、海藻酸钠(SA)为原料,戊二醛(GA)为交联剂,在酸性缓释条件下制备壳聚糖-海藻酸钠(CS-SA)天然生物质水凝胶.实验结果表明,相比于外凝胶法,内凝胶法可促使缓慢成胶,避免形成不均匀的沉淀物.进一步,通过系列实验发现CS/SA=1/3(即CS 0.2 g,SA 0.6 g),交联剂戊二醛GA用量 0.3 mL,并采用冷冻干燥的方式,可制备得到内部结构疏松多孔的CS-SA水凝胶.FTIR以及SEM结果表明CS-SA合成成功且具有多孔疏松的微观结构,该特性可潜在提高CS-SA对溶液中三价铬的吸附效能又利于其后期降解用于制革复鞣填充.基于此,本论文提供的CS-SA天然生物质水凝胶的制备方法,可潜在实现集Cr(III)治理-复鞣填充一体化,满足吸附剂-吸附质最大资源化利用.
猜你喜欢
包装工程(2022年15期)2022-08-23
陕西农业科学(2022年6期)2022-08-10
辽宁化工(2022年5期)2022-05-28
中国药学药品知识仓库(2022年7期)2022-05-10
包装工程(2022年5期)2022-03-21
能源工程(2021年6期)2022-01-06
粘接(2021年2期)2021-06-10
科技创新导报(2017年34期)2018-06-05
求知导刊(2016年16期)2016-07-28
山西果树(2014年3期)2014-07-15
