
多重生物信息分析预测免疫相关基因FN1为干燥综合征发病核心基因
2022-02-13 10:41吉林省白城医学高等专科学校白城137000
中国免疫学杂志 2022年23期
薛 娜 肖 莹 董 晶 孙 伟 (吉林省白城医学高等专科学校,白城137000)
干燥综合征(Sjögren's syndrome,SS)是一种全身性自身免疫病,主要病理生理学改变是外泌腺淋巴细胞浸润引起的与SS 相关的眼睛和口腔干燥。SS 是第二常见的慢性自身免疫性疾病,其发病具有性别倾向,主要集中于50岁左右的女性患者[1-2]。SS分为原发性和继发性两种类型,目前尚无单一的实验室检验或检查可确诊此疾病[3]。2016年ACR/EU‑LAR 指南主要通过结合唾液腺活检的抗Ro/SSA 阳性及眼部和口腔干燥症状分类[4]。此外SS 还会导致患者出现腺外表现,影响多种器官,包括肺、皮肤、关节、神经系统和肾脏,进而加重患者的生活负担和病死率[5]。目前SS 病因不明,因此无特效治疗药物,临床治疗主要以缓解症状、延缓病情发展为主要目的。因此本研究从基因水平探究SS 的发病机制,应用多重生物信息分析方法试图寻找到与SS发病过程最相关的核心基因及关键通路,为SS的快速诊断和治疗提供理论依据。
1 材料与方法
1.1 基因数据来源 选取GEO数据库中SS相关基因原始数据GSE97614,共包含9 例SS 患者及3 例体检者的基因检测结果,从GPL6244 平台下载入组患者的基本状况及微矩阵原始数据[6]。全部基因应用GraphPad Prism 7.0绘制火山图进行可视化处理,应用基因热图在TBtools软件上进行聚类分析。
1.2 数据校准及筛选 应用R 语言limma 数据包对基因微阵列原始数据进行均一化处理,筛除表达量过高或过低的原始数据。应用GEO2R 算法对全部基因进行处理,并依照P<0.05和差异表达倍数>2的标准进行筛选,从而获得差异上调表达和差异下调表达的基因。
1.3 DAVID 数据富集分析 应用DAVID 在线分析软件分别对差异上调和差异下调及全部差异表达基因(differentially expressed genes,DEGs)进行基因本体(GO)分析及KEGG 信号通路分析。其中GO 分析包括细胞组分、分子功能及生物过程。全部富集分析筛选标准为基因数≥2个及P<0.05。对DEGs富集结果进行R语言数据处理及气泡图可视化呈现。
1.4 蛋白互作(PPI)网络分析 应用STRING 在线分析软件进行PPI 网络制作,全部DEGs输入“Multi‑ple proteins”对话框,物种选择为“homo sapiens”,同时获得可视化处理后的PPI网络图及基因间的具体联系。
1.5 核心基因预测 将PPI 网络图提供的数据信息输入Cytoscape 软件进行可视化处理。通过Cyto‑scape 内置插件cytohubba 对全部基因联系进行分析和计算,选择“Degree”评分准则对全部DEGs按照分数依次降序排列,最终筛选出与SS发病最为相关的核心基因。
1.6 统计学方法 所有原始数据采用R 语言软件进行分析。DEGs 筛选标准为差异倍数≥2 及P<0.05。KEGG 信号通路富集基因最少为2 且P<0.05。PPI筛选标准为基因间联系>0.4。
2 结果
2.1 原始数据处理 GSE97614 共包含12 例患者样本,全部患者均为女性,依照ACR/EULAR 指南诊断为原发性SS。其中9 例SS 患者平均年龄为58 岁(24~75 岁),对照组3 例患者平均年龄为52 岁(20~70 岁)。SS 患者具体服药情况不详。GSE97614 共包含49 395 个编码基因的检测结果,数据样本的均值-方差趋势(图1A)、UMAP 图(图1B)、表达密度(图1C)、矫正P值(图1D)、t值(图1E)显示基因检测结果具有可分析性。

图1 GSE97614原始数据一般概况Fig.1 General overview of GSE97614
2.2 差异基因矫正 检测12例样本中每例样本的49 395 个编码基因数据,结果显示原始数据结果均一性较差(图2A);应用R 语言limma 包进行数据均一化处理,筛除离散度较大的数据(表达过高或表达过低),校准后的数据均一性较好(图2B)。

图2 差异基因矫正Fig.2 DEGs correction
2.3 差异基因筛选及可视化处理 共筛选出96个差异上调表达和65 个下调表达的基因。对全部DEGs 进行火山图可视化处理结果见图3A。对全部DEGs 进行聚类分析,应用基因热图进行可视化处理,结果显示全部DEGs具有可聚类性(图3B)。
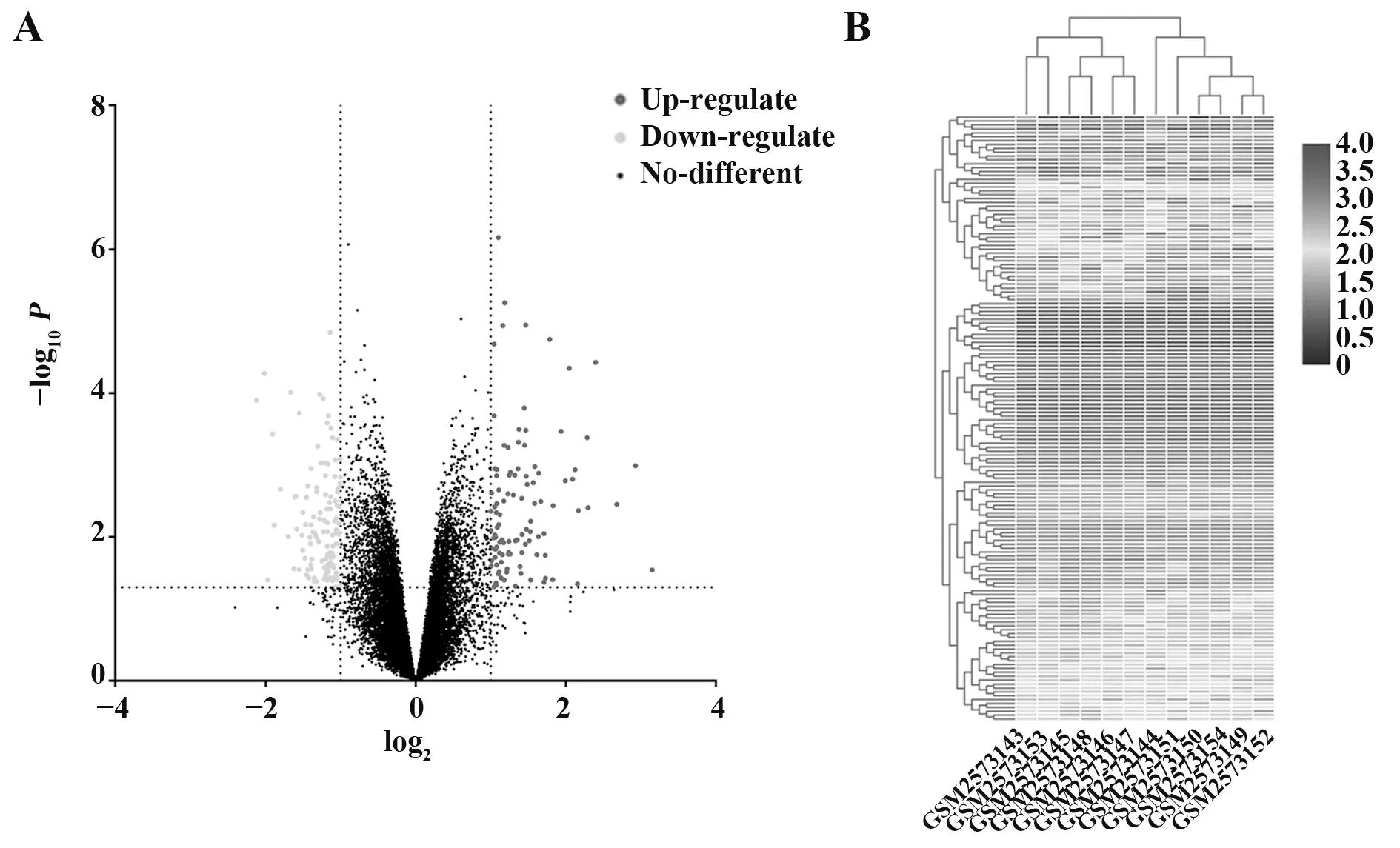
图3 差异基因可视化处理Fig.3 Visualization of DEGs
2.4 差异基因的GO 分析 对差异上调表达基因进行GO 分析显示差异基因在细胞组成主要富集于细胞外间隙,生物过程主要富集于细胞外基质,分子功能主要富集于细胞因子活性。差异下调表达基因在细胞组成主要富集于质膜的组成成分,生物过程主要富集于细胞对激素刺激的反应,分子功能主要富集于整合素结合。全部DEGs 在细胞组成主要富集于细胞外间隙,生物过程主要富集于细胞外基质,分子功能主要富集于细胞因子活性,和差异上调表达基因的GO分析一致,说明差异上调表达基因可能在SS的发病过程中发挥更重要的作用(图4)。

图4 差异基因的GO分析Fig.4 GO analysis of DEGs
2.5 差异基因的KEGG 分析 对差异上调表达基因进行KEGG分析显示差异基因主要富集于补体和凝血级联通路、细胞外基质通路及黏着斑通路。对差异下调表达基因进行KEGG分析显示差异基因主要富集于干细胞多能性信号通路、雌激素信号通路及MAPK信号通路(图5)。

图5 差异基因的KEGG分析Fig.5 KEGG analysis of DEGs
2.6 PPI网络 STRING在线分析软件对全部161个DEGs 进行PPI 网络分析,结果显示共有141 个基因之间存在相关性联系,联系线路为224条,每个基因间的连接程度评分为3.18,平均局部聚类系数为0.401,PPI富集P值<1.0e-16(图6)。
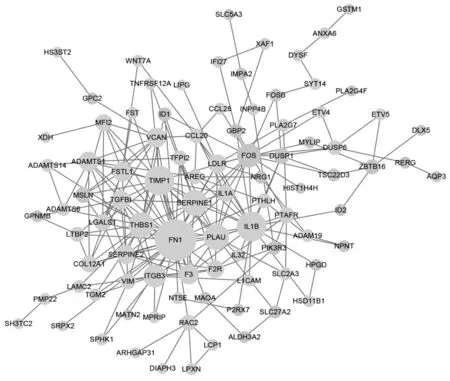
图6 差异基因PPI网络Fig.6 PPI network of DEGs
2.7 SS发病核心基因预测 应用Cytoscape对蛋白网络连接数据进行可视化处理,cytohubba 内置应用程序计算出FN1(32分)、IL1B(20分)、TIMP1(20分)、THBS1(16分)、PLAU(16分)、SERPINE1(16分)、FOS(14 分)、ITGB3(13 分)、VCAN(10 分)、ADAMTS1(10 分)为前10 位发病核心基因。对10 个核心基因进行聚类分析显示基因间具有可聚类性(图7)。

图7 核心基因预测Fig.7 Prediction of hub genes
3 讨论
SS 是一种慢性自身免疫病,其典型的病理学改变是泪腺和唾液腺受累,导致眼睛和口干燥。SS 可累积到全身多个器官,包括肺、肾和中枢神经系统,加重患者的生活负担及病死率[7]。目前针对SS 的具体发病机制尚不明确,因此无特效治疗药物。西医以激素及免疫抑制剂为主,并针对症状选择人工泪液、人工唾液进行对症支持治疗。中医多采用以补益脾胃、益气养阴为主的辨证治疗[8]。目前针对SS的诊断也是临床的难点,由于SS患者早期缺乏特异性临床表现,临床也缺乏特异性和敏感性高的血清诊疗标志物,常导致SS治疗时机延误。因此新的快速的血清学生物诊疗标志物可提供SS 的早期诊断,更有可能成为治疗SS的生物靶点。本研究主要对既往SS疾病基因测序结果进行深入分析,试图找到SS快速诊疗的标志物及潜在治疗靶点。
纤维连接蛋白(fibronectin,FN)是脊椎动物的一种470~500 kD 的糖蛋白,对动物的发育、器官生长、细胞黏附、细胞迁移及止血等正常过程发挥重要作用[9]。FN分为3种亚型,包括FN1、FN2和FN3。FN 一般有两种来源:血浆FN 由肝细胞合成并分泌到血液中;细胞FN 由细胞局部分泌。FN 需要被组装成细胞外基质的原纤维发挥作用。目前关于FN的组装机制主要在细胞层面进行研究,涉及FN 与细胞表面分子结合,招募额外的FN 分子形成不溶性原纤维。组装开始于细胞黏附部位,生长的原纤维可向内部转移[10]。FN1分子广泛分布于健康的细胞膜、固有层、血管结构、神经和平滑肌细胞层[11]。其是细胞迁移和转移的重要调节因子,据报道,FN1参与人类皮肤鳞状细胞癌的发生[12]。FN1在甲状腺癌中表达上调,下调其表达可抑制甲状腺癌细胞的增殖、侵袭和迁移,提示FN1 可能参与甲状腺癌的发生[13]。FN1 通过激活PI3K 促进细胞增殖,抑制细胞凋亡[14]。在肝细胞癌中发现FN 蛋白呈过表达趋势[15]。胃肠道和头颈部癌症患者血浆FN1 水平明显升高[16]。在非恶性疾病中,特别是在血栓形成、止血过程、血管疾病和血小板功能方面,FN 的确切作用仍不明确。
目前针对FN1 在SS 发病机制中的作用尚未得到广泛关注,国内外仅有2 篇报道。JIANG 等[17]在2021 年的研究发现沙参麦冬汤可能对SS 的治疗发挥积极作用,其通过生物信息学分析发现,沙参麦冬汤可能通过靶向结合FN1 治疗SS。其具体机制可能与沙参麦冬汤改善FN1 和MMP-9 对唾液腺泡和基底膜结构的破坏、降低FN1在SS中的促炎症反应有关。MONA 等[18]2020 年的体内实验发现,具有潜在免疫调节及治疗作用的骨髓间充质干细胞在与唾液腺细胞进行共培养时,可分化为唾液祖细胞,取代病变的唾液腺细胞从而起到治疗SS 的作用。
本文通过生物信息分析方法,首先对GSE97614基因检测结果进行了均一化处理,使得后续生物信息分析结果更加准确。然后利用GO分析、KEGG 信号通路分析,PPI 网络分析及cytoHubba 核心发病基因计算最终发现FN1 是SS 发病的关键基因。本文并不是第一篇针对SS 发病核心基因预测的文章。目前有4 篇针对SS 发病基因的生物信息分析文献。郭俊恺等[19]分析GSE23117 和GSE127952 的基因表达谱发现CXCL9 可能与SS 发病相关。蔡鑫等[20]分析GSE127952 的基因表达谱发现STAT1 可能与SS发病相关。徐华等[21]分析GSE7451、GSE127952 和GSE40611的基因表达谱发现STAT1可能与SS发病相关。梁江等[22]分析GSE23117、GSE07451、GSE40611、GSE84844 的基因表达谱发现BAFF 可能与SS 发病相关。目前尚无研究分析GSE97614 数据,而本文的分析结果也与既往研究发现的核心发病基因不完全相同,提示SS发病机制的复杂性及多样性。
FN1 是上皮细胞-间质细胞转化相关基因,在肿瘤相关研究中被关注较多。近年来,越来越多的研究表明FN1 也是参与免疫浸润的免疫相关基因。FN1编码FN并储存于血浆和细胞外基质,在炎症组织募集巨噬细胞过程中,FN1 呈现高度上调趋势。与此同时,FN1在体外也可作为白细胞迁移的基质,促进组织中的T 细胞聚集,增强局部对感染或癌症的免疫力[23]。免疫浸润分析研究显示免疫浸润水平与FN1表达水平相关。FN1参与调控NKp46受体介导的天然杀伤细胞产生IFN-γ,进而影响细胞内免疫。FN1 参与许多与免疫相关的信号通路,如趋化因子信号通路、细胞因子受体相互作用、自然杀伤细胞介导的细胞毒性和T 细胞受体信号通路。FN1 表达水平的增加与M2 巨噬细胞和静息CD4+T细胞的比例呈正相关,但与滤泡辅助性T 细胞和CD8+T细胞比例呈负相关[24]。SS是一种自身免疫性疾病,随着FN1 参与体内免疫应答过程的深入研究,其在SS发病机制中的作用亦会被逐渐挖掘。
综上,本研究通过对SS基因数据进行生物信息学分析发现免疫相关基因FN1 可能通过改善免疫调节诱发SS。FN1 可能作为SS 快速诊断的血清标志物及潜在的药物干预靶点。未来有关FN1 介导免疫浸润、免疫细胞的募集及其在SS发病机制中的作用可能成为重点研究方向。
猜你喜欢
China Report Asean(2022年8期)2022-09-02
世界科学技术-中医药现代化(2022年3期)2022-08-22
师道·教研(2022年1期)2022-03-12
物联网技术(2020年12期)2021-01-27
海洋信息技术与应用(2020年1期)2020-06-11
心电与循环(2020年1期)2020-02-27
传媒评论(2019年4期)2019-07-13
汽车零部件(2017年4期)2017-07-12
江苏农业科学(2017年5期)2017-04-15
湖北农业科学(2014年3期)2014-07-21
