
不同进样方式对气相色谱法测定液化石油气组成结果的影响
2022-02-01 10:20刘荣吴宇林杨杰涂瀚匀
化学分析计量 2022年12期
刘荣,吴宇,林杨杰,涂瀚匀
(1. 四川省产品质量监督检验检测院,成都 610100; 2. 成都产品质量检验研究院有限责任公司,成都 610100;3. 暨南大学基础医学与公共卫生学院,广州 510632)
液化石油气是在石油炼制过程中由多种低沸点烃类组成的混合物,主要来源于炼油厂二次加工装置和油田伴生气,在常温常压下为气体,只有在加压或降温的条件下,才变成液体,其主要成分是丙烷、丙烯、丁烷、丁烯[1-8]。由于液化石油气特殊的气液共存性状[9-10],对其组成的准确测定是评价液化石油气质量好坏的一项重要指标[11-13]。
GB/T 11174—2011《液化石油气》[14]中规定液化石油气组成的仲裁分析方法为NB/SH/T 0230—2019《液化石油气组成的测定 气相色谱法》[15],该方法对液化石油气色谱分析的进样方式未进行详尽的规定。笔者研究发现进样方式的不同对液化石油气组成的测定有着不可忽视的影响,而经文献调研未见关于不同进样方式的相关报道。笔者采用双FID 气相色谱测定液化石油气的组成,选择了当前主要应用的三种进样方式对液化石油气组成测定结果进行对比研究,验证了不同进样方式在不同条件下的可行性,为液化石油气组成的准确测定提供了参考依据,为国家和行业标准的完善和制修订提供了理论支撑,具有重要意义。
1 实验部分
1.1 主要仪器与试剂
气相色谱仪:Agilent 8890 型,配置双FID 检测器、双六通阀,安捷伦科技(中国)有限公司。
闪蒸进样器:(1)Agilent G3535 A型(减压阀型),安捷伦科技(中国)有限公司;(2)盘管式,大庆市日上仪器制造有限公司。
高压液体进样阀:Agilent G3505 A 型,安捷伦科技(中国)有限公司。
丙烷、丙烯、异丁烷、异丁烯、正丁烷、正丁烯、反-2-丁烯、顺-2-丁烯、异戊烷、1-戊烯、二甲醚、甲基叔丁基醚、甲醇标准溶液:标准物质编号BW(DT)0103,体积分数分别为55.547 3%、0.112%、1.94%、20.1%、20.57%、0.118%、0.111%、0.111%、0.098 6%、0.102%、1.17%、0.01%、0.010 1%,大连大特气体有限公司。
高纯氦气、高纯氢气、高纯氮气:纯度(体积分数)均不低于99.999%,四川文茂气体有限公司。
压缩空气:四川文茂气体有限公司。
1.2 仪器工作条件
色谱柱:(1)Agilent HP-AL/M 型毛细管柱(30 m×0.53 mm,15 μm),(2)Agilent Lowox 型毛细管柱(10 m×0.53 mm,10 μm),安捷伦科技(中国)有限公司;进样口温度:200 ℃;柱温:程序升温;柱升温程序:初始温度为90 ℃,保持1 min,以6 ℃/min 升温至180 ℃;载气:高纯氦,流量为4.5 mL/min;进样方式:闪蒸或液体阀进样,分流比为40∶1;检测器温度:300 ℃,燃气:高纯氢气,流量为30 mL/min;助燃气:压缩空气,流量为400 mL/min;尾吹气:高纯氮气,流量为25 mL/min。
1.3 进样方式
1.3.1 减压式闪蒸进样
样品瓶倒置连接汽化系统,汽化系统通过专用减压阀将样品压力瞬间从几个MPa 降至低于0.1 MPa,液化石油气样品瞬间汽化,在经钝化的减压阀和传输管线上进行加热,以防止汽化后样品重新冷凝。汽化后的样品经气体六通阀进入气相色谱进行检测。
1.3.2 盘管闪蒸进样
汽化系统由一定长度的内壁钝化的不锈钢盘管组成,样品瓶倒置,液化石油气样品进入闪蒸仪盘管,盘管被加热到80~100 ℃,汽化后经气体六通阀进入气相色谱进行检测。
1.3.3 液体阀进样
样品瓶倒置连接液体阀定量环,高压液体阀提供高阻尼,液化石油气样品在高阻尼情况下完全液化,通过连接定量环出口的进样针直接导入气相色谱仪进样口完成进样。
2 结果与讨论
2.1 不同进样方式下的烃类校正因子
取各标准溶液,应用三种不同的进样方式连续进样5次,按照式(1)计算各烃类组分校正因子,取平均值:

式中:fVi——校正样品中组分i校正因子;
VTi——校正样品中组分i体积分数标称值,%;
VTs——校正样品中基准组分s 体积分数标称值,%;
ATi——校正样品中组分i色谱峰面积;
ATs——校正样品中基准组分s色谱峰面积。
由式(1)分别得到一组烃类体积分数校正因子,获得的校正因子分别与NB/SH/T 0230—2019 给定的典型体积分数校正因子进行比较,结果列于表1。由表1 可知,减压式闪蒸进样和液体阀进样两种方式计算所得烃类体积分数校正因子与给定的典型体积分数校正因子非常接近,相对误差均低于5%。盘管闪蒸进样所得烃类体积分数校正因子与前两种进样方式相比相对误差略偏大,为-10.61%~6.38%。
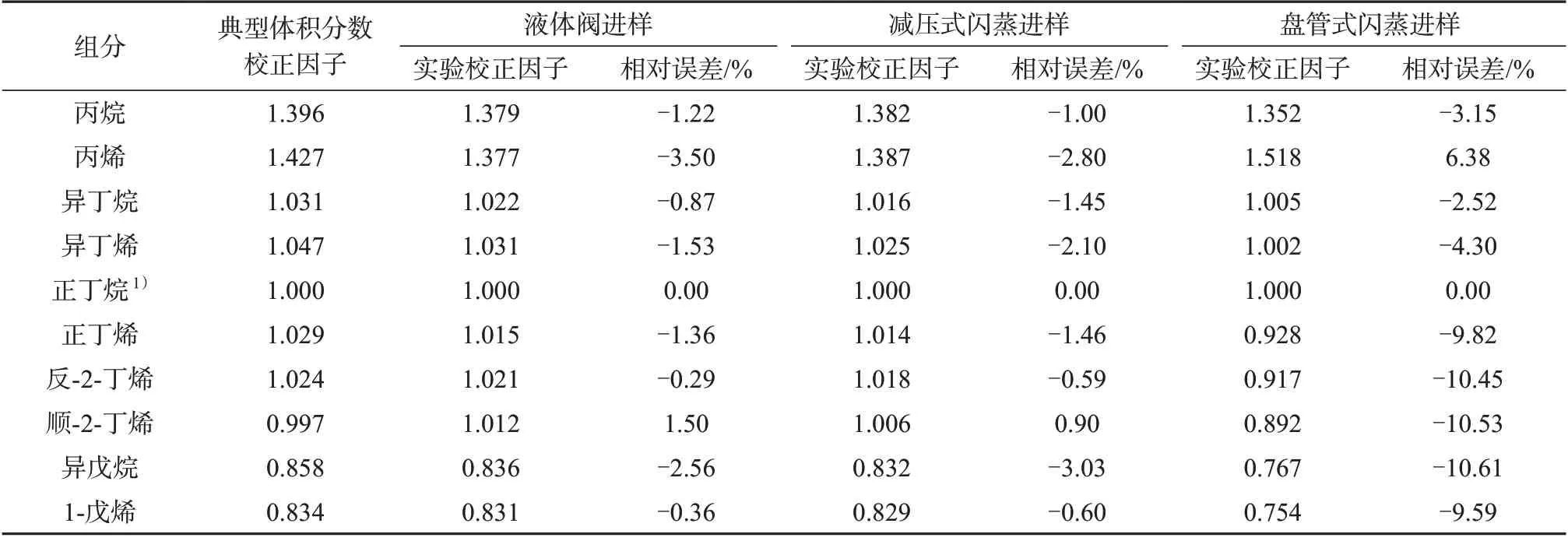
表1 烃类体积分数校正因子
2.2 进样方式对测定结果的影响
配制组分浓度相同、充装压力分别为1 MPa 和2 MPa 的2 瓶液化石油气样品,在规定的实验条件下,应用以上三种进样方式分别对这两个液化石油气样品连续进样测定5 次,对测定结果的准确度和精密度进行分析。校正曲线方程为式(2),据此计算各个氧化物组分的体积分数,然后按式(3)用差减校正归一方式计算各烃类组分的体积分数:
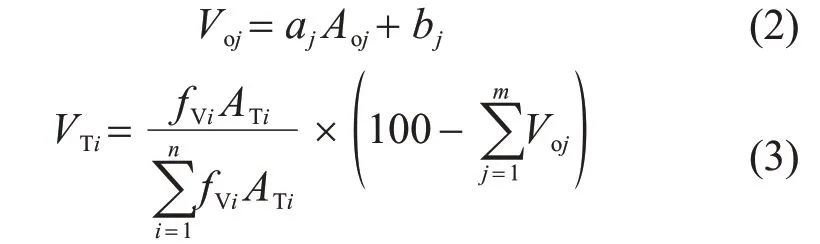
式中:Voj——含氧化合物组分j体积分数,%;
Aoj——含氧化合物组分j色谱峰面积;
aj,bj——含氧化合物组分j校正曲线的斜率和截距;
VTi——待测烃类组分i体积分数,%;
fVi——待测烃类组分i校正因子;
ATi——待测烃类组分i色谱峰面积;
n——待测烃类组分数量;
m——待测含氧化合物组分数量。
表2~表7 分别为进样压力1 MPa 和2 MPa 下三种进样方式的测定结果。由表2~表4可以看出,进样压力1 MPa 时,三种进样方式表现各不相同。以液体进样阀进样所测得的13 个组分测定结果的相对标准偏差均小于1%,回收率仅有3个含量高的组分(正丁烷、异丁烷、丙烷)分别达到100.7%、102.6%和91.2%,表明该进样方式精密度良好,但准确度仅有含量较高组分表现较好,其它组分准确度较差。减压式闪蒸进样所测得的各组分测定结果的相对标准偏差均小于1%,回收率为96.0%~108.0%,表明该种进样方式所测结果精密度和准确度均良好。盘管式闪蒸进样所测得结果的RSD有3个组分达到了1%以上,其余均小于1%,表明精密度良好;回收率为8%~211%,准确度差。

表2 1 MPa进样压力下液体阀进样测定结果
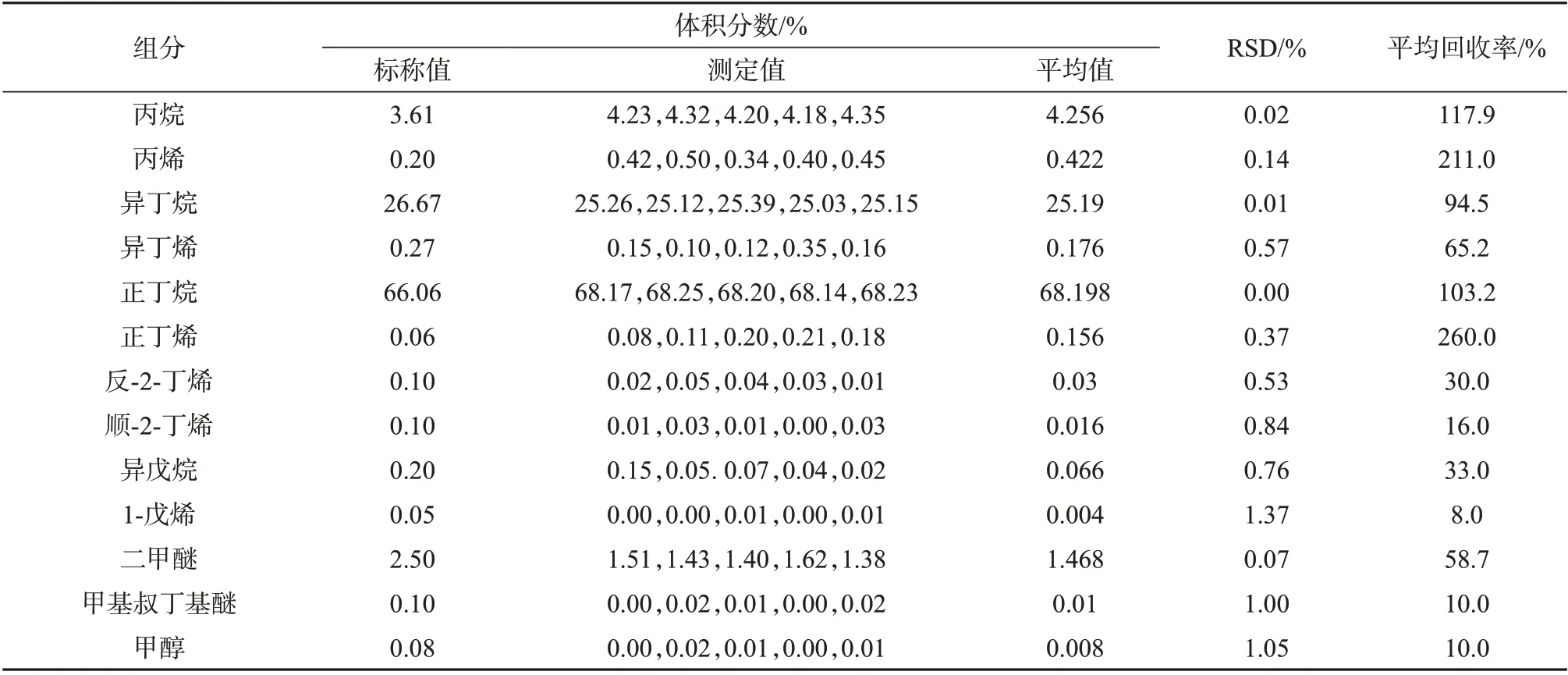
表4 1 MPa进样压力下盘管闪蒸进样测定结果
由表5~表7 可以看出,进样压力2 MPa 时,三种进样方式亦有差别。以液体进样阀进样所测得的各组分测定结果的相对标准偏差均小于0.5%,回收率为96.0%~106.0%,表明该种进样方式所测结果的精密度和准确度良好。减压式闪蒸进样所测得的各组分测定结果的相对标准偏差均小于0.5%,回收率为90.0%~113.0%,表明该种进样方式所测结果精密度和准确度良好。盘管闪蒸进样所测得结果的RSD 均小于0.5%,回收率为68.0%~135.0%,表明该进样方式精密度较好,但准确度较差。
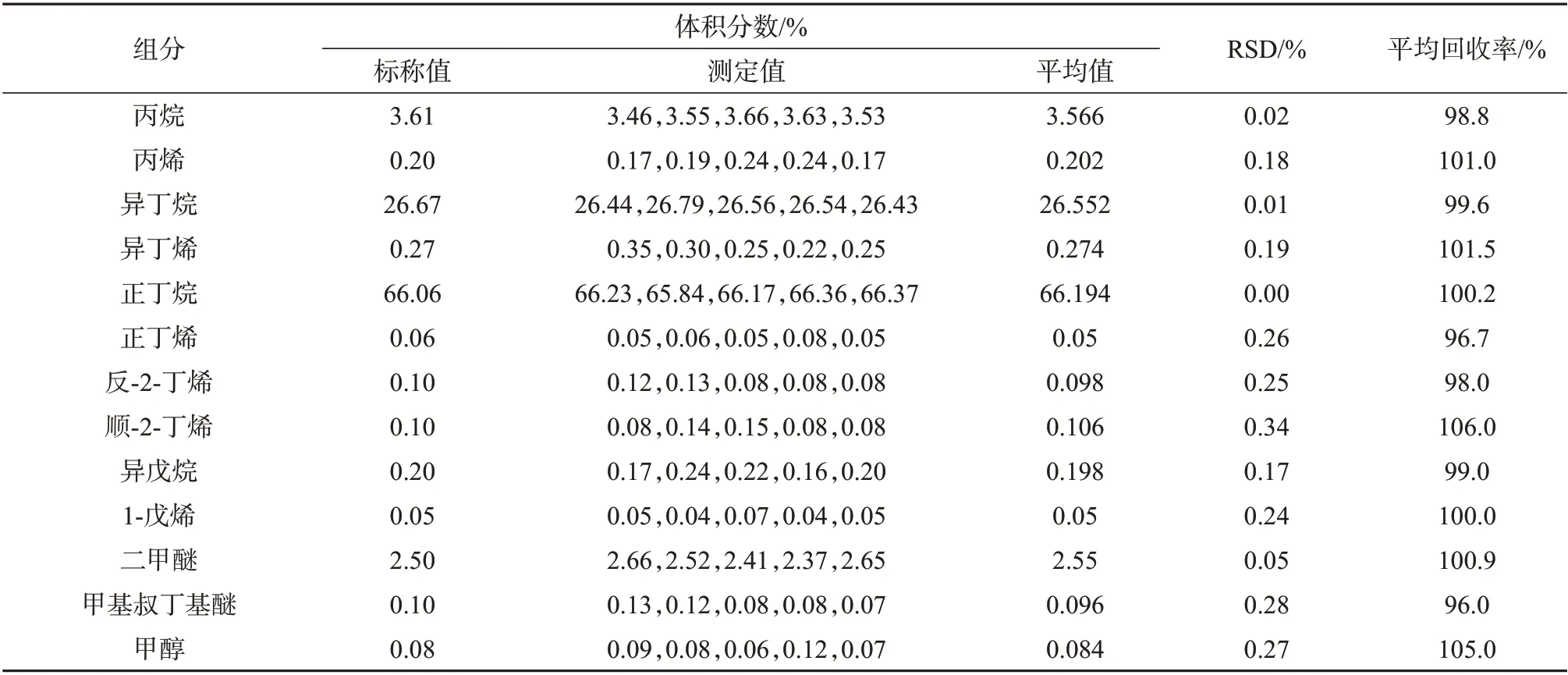
表5 2 MPa进样压力下液体阀进样测定结果
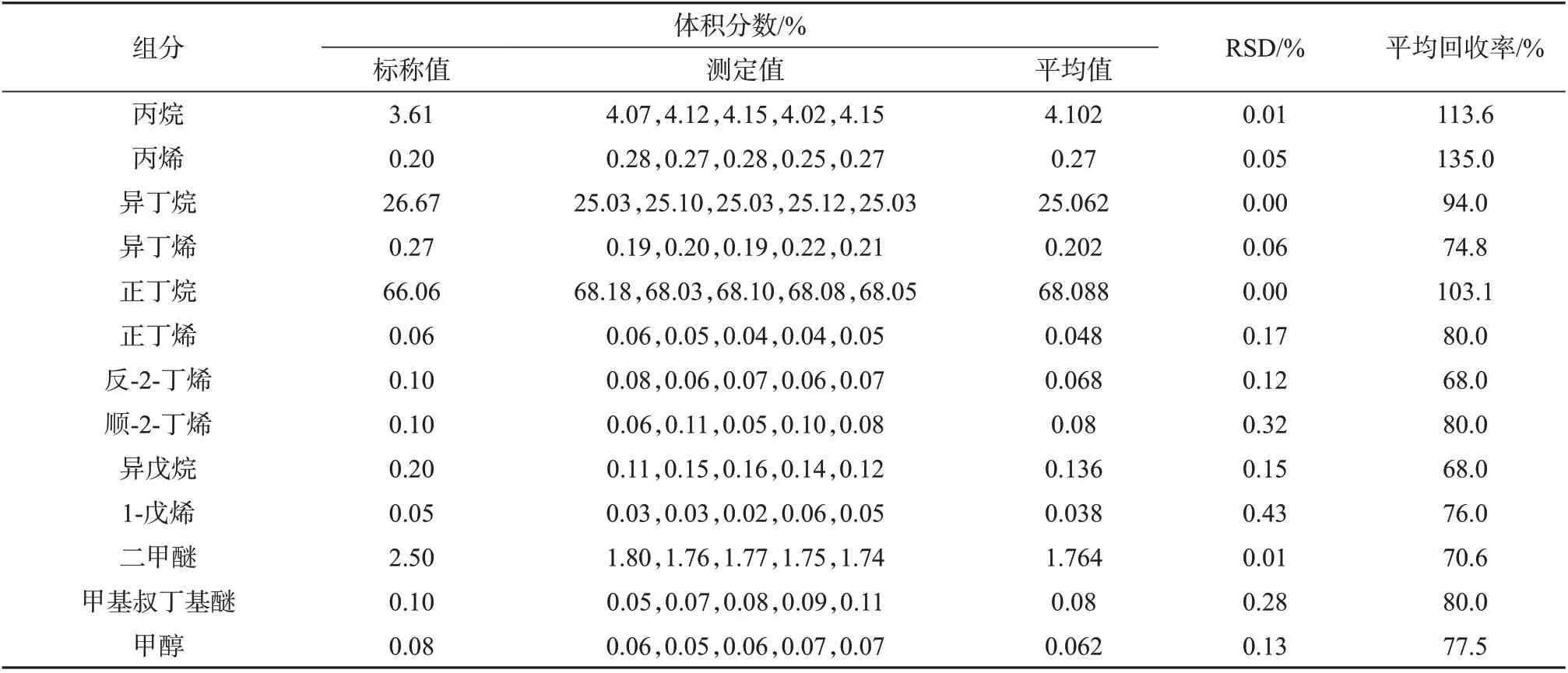
表7 2 MPa进样压力下盘管闪蒸进样测定结果
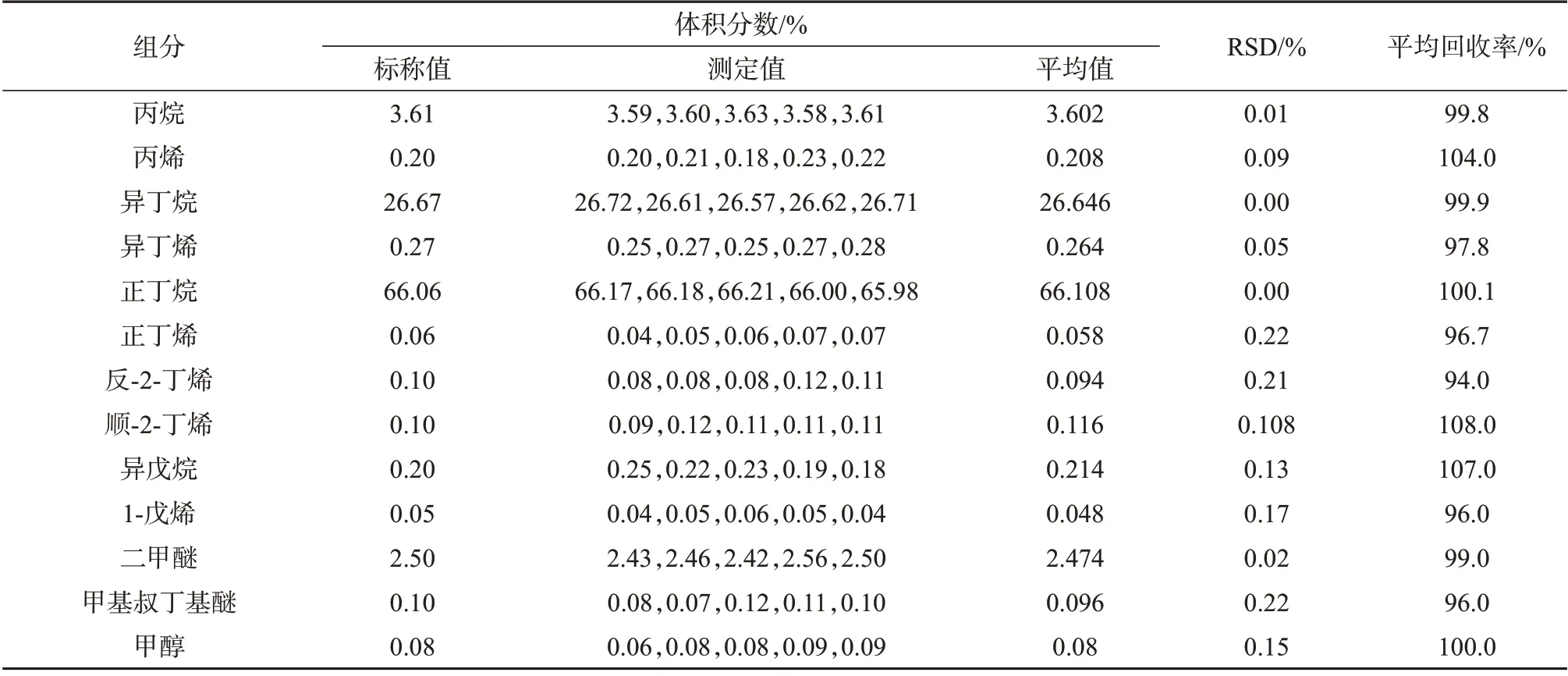
表3 1 MPa进样压力下减压式闪蒸进样测定结果
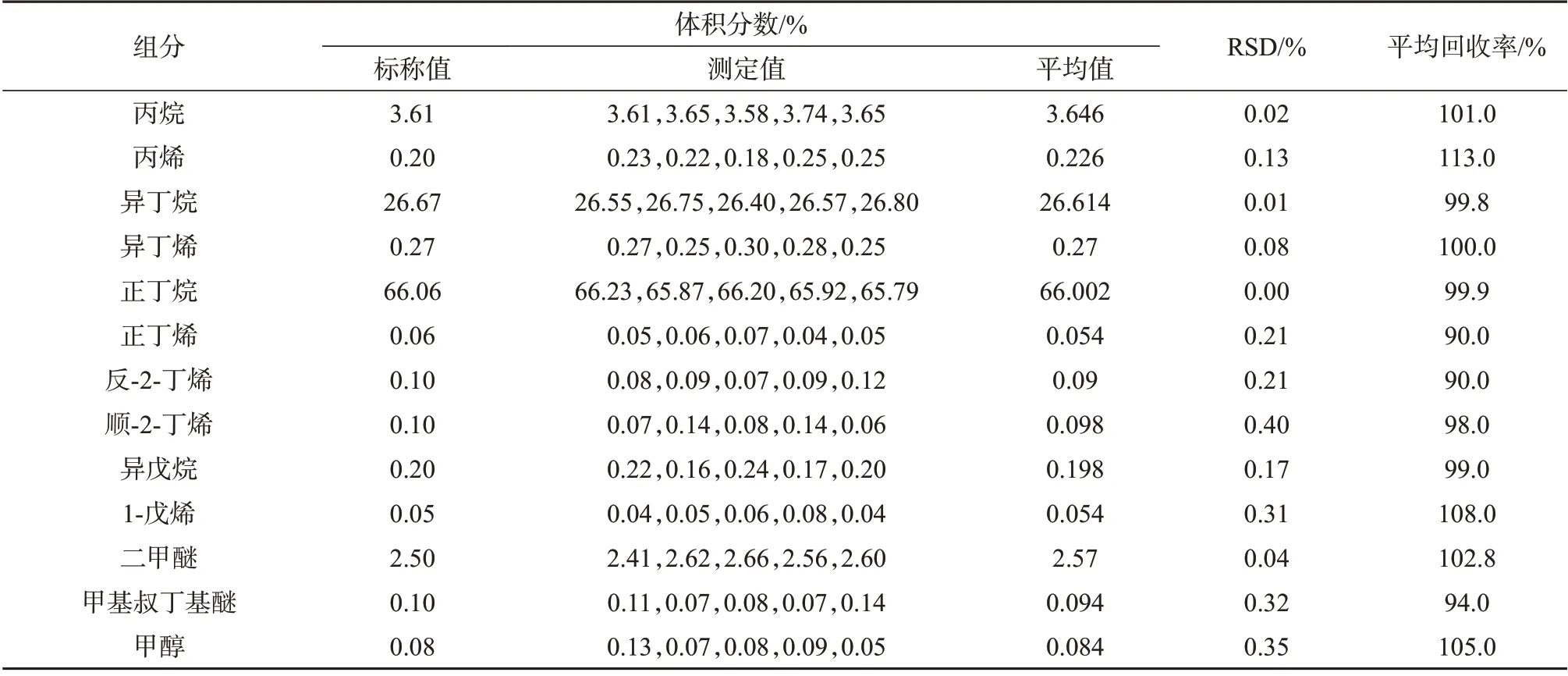
表6 2 MPa进样压力下减压式闪蒸进样测定结果
从以上试验数据可以看出,在进样压力不大于1 MPa 时,仅有减压式闪蒸进样方式测得结果具有良好的精密度和准确度;液体阀进样所测得的结果对于含量较高的组分具有较好的精密度和准确度;而盘管式闪蒸进样准确度达不到实验的要求。在进样压力高于1 MPa 时,减压式闪蒸进样和液体进样阀进样两种方式均具有较好的精密度和准确度;盘管式闪蒸进样所测结果的准确度较差。究其原因,由于液体阀进样需要较高的进样压力才能保证液化石油气样品充分均匀地液化,如进样压力较低,则可能造成进入色谱柱的组分不均匀,使所测结果特别是含量低的组分测试结果存在较大的偏离。而盘管式闪蒸进样会导致低沸点的组分先于高沸点的组分蒸发,造成分馏,虽然液化气在通过盘管后样品都已转变为气体,但是汽化后的样品分子比例已经与分馏之前有所不同,造成组分浓度的改变。
2.3 样品中含有C2时进样方式的影响
配制压力充足含有一定量C2组分的液化石油气样品,分别采用减压式闪蒸和液体阀连续进样两次,组分测定结果列于表8 和表9。从表8 和表9 可以看出,两种进样方式两次进样的所有结果均满足液化石油气组成仲裁分析方法NB/SH/T 0230—2019给出的重复性要求。对减压式闪蒸进样,所测结果的相对误差为-4.17%~6.25%,其中C2组分的相对误差为0.40%~0.79%,准确度较好。对液体阀进样,所测结果的相对误差为-10.08%~9.38%,其中C2组分的相对误差达到-10%左右,较之减压式闪蒸进样,尤其是C2组分的准确性要偏低一些。原因在于若液化石油气含有一定量的乙烷,乙烷沸点较低,液体阀不易将乙烷完全液化,造成测定结果偏低。由于盘管式闪蒸进样对于含有一定量C2的液化石油气具有不稳定性,此处不再进行比较。
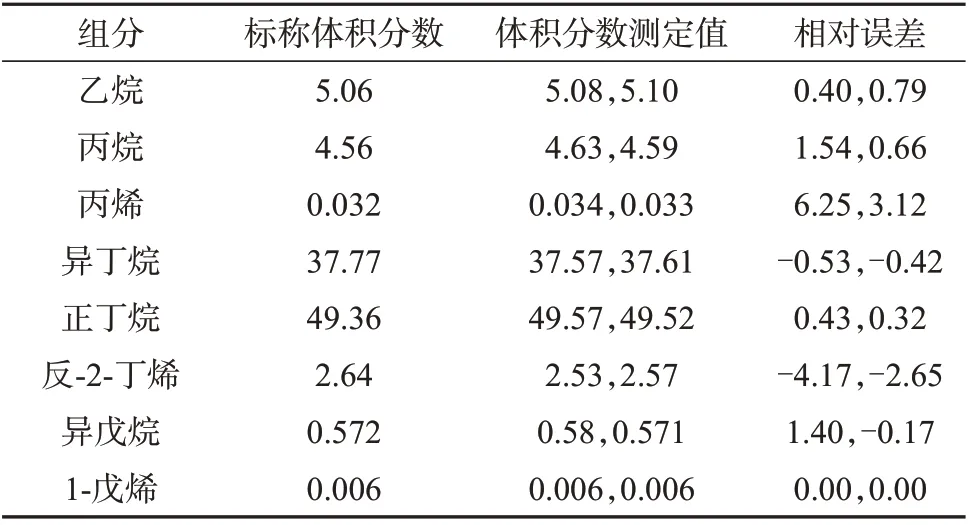
表8 减压式闪蒸进样方式测定结果 %
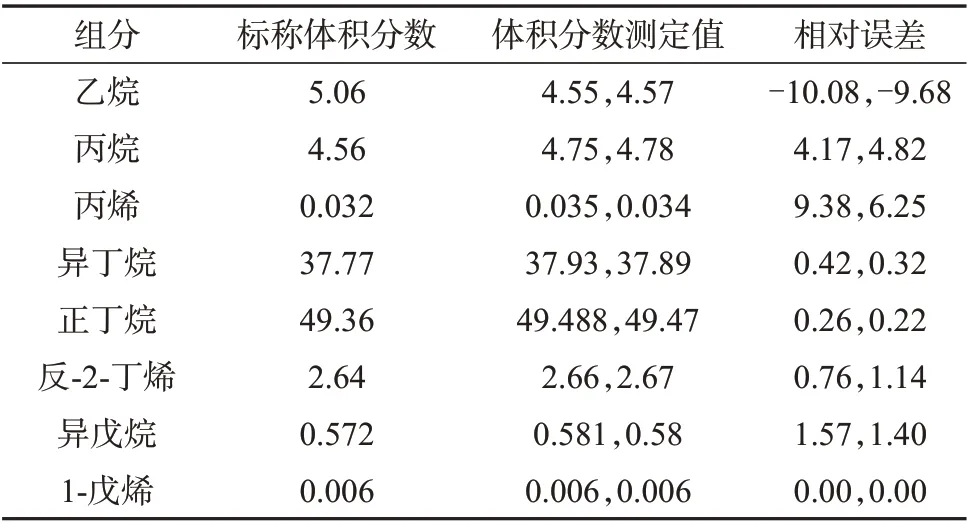
表9 液体阀进样方式测定结果 %
3 结语
对液化石油气组成测定所选用的不同进样方式的考察结果表明,减压式闪蒸和液体阀进样两种方式所测烃类体积分数校正因子与NB/SH/T 0230—2019 中给定的典型体积分数校正因子非常接近。在进样压力低于1 MPa 时,仅减压式闪蒸进样所测结果具有较好的精密度和准确度。进样压力高于1 MPa时,减压式闪蒸和液体阀两种进样方式所测结果均能够达到较好的精密度和准确度。对于含有一定量C2的液化石油气,宜采用减压式闪蒸进样,而液体阀进样准确度偏低,盘管式闪蒸进样具有不稳定性,不宜采用。
猜你喜欢
中国特种设备安全(2022年6期)2022-09-20
大连理工大学学报(2021年6期)2021-11-29
世界有色金属(2021年12期)2021-11-02
中国特种设备安全(2019年11期)2020-01-16
世界有色金属(2018年8期)2018-06-28
世界有色金属(2018年10期)2018-01-30
中国交通信息化(2016年5期)2016-06-06
智能城市(2016年2期)2016-02-09
现代企业(2015年1期)2015-02-28
