
19例cblC型甲基丙二酸血症患儿临床资料及基因型分析
2022-01-28 08:00魏晨曦赵婉晴张亚男
临床荟萃 2022年1期
魏晨曦,赵婉晴,张亚男
(河北医科大学第二医院 儿科,河北 石家庄 050000)
甲基丙二酸血症(methylmalonic acidemia, MMA)是由于甲基丙二酰辅酶A变位酶缺陷或其辅酶钴胺素(维生素B12)代谢缺陷导致的一种常染色体隐性遗传病[1]。根据缺陷部位的不同可分成甲基丙二酰辅酶A变位酶缺陷(mut型)和辅酶钴胺素代谢障碍两大类。根据血同型半胱氨酸是否增高,可分成单纯型MMA和MMA合并同型半胱氨酸血症[2]。cblC型MMA是MMA合并同型半胱氨酸血症中最常见的一种类型,由于MMACHC基因突变导致辅酶钴胺素代谢障碍,造成甲基丙二酸异常蓄积,血同型半胱氨酸增高等变化,最终导致多脏器功能受损,如神经系统(肌张力低下、癫痫发作、眼球震颤、双眼凝视等)、心血管系统(扩张型心肌病、肺动脉高压、肺心病等)、血液系统(贫血、粒细胞减少等)、内分泌系统(高氨血症[3])、眼部疾病(严重时会导致黄斑病变和色素性视网膜病变等[4])。随着串联质谱技术在新生儿中的广泛应用,我们对该病的认识在逐渐加深。本研究对河北医科大学第二医院19例cblC型MMA患儿的临床资料进行了回顾性分析,以增加对该病的认识,并为其诊断、治疗提供依据。
1 资料与方法
1.1病例选择 收集2015-2020年于河北医科大学第二医院小儿内分泌与生长发育科诊治的cblC型MMA患儿19例,男10例, 女9例。8例通过新生儿筛查确诊。本研究经过河北医科大学第二医院伦理委员会批准, 所有检查均获得患儿及家属的同意。
1.2方法 回顾性分析患儿的临床资料,包括:①临床表现:发育落后、注意力不集中、喂养困难、反应差等;②一般检查:血、尿常规,肝、肾功能,血气分析,血糖,血氨,血乳酸;③氨基酸及酰基肉碱检测丙酰肉碱(propionyl carnitine, C3)、乙酰肉碱(acetyl carnitine, C2)、C3/C2、C3/游离肉碱(free carnitine, C0)、蛋氨酸(methionine, Met);④尿有机酸测定:甲基丙二酸、甲基枸橼酸-4;⑤血同型半胱氨酸(homocysteine, HCY);⑥影像学检查:脑电图、心脏超声、头颅磁共振成像(magnetic resonance imaging, MRI);⑦MMACHC基因突变检测。其中尿液有机酸综合分析报告和氨基酸和酰基肉碱谱分析报告采用串联质谱分析方法,由金域检验提供,基因分析报告由北京迈基诺医学研究所提供。
2 结 果
2.1临床表现 19例cblC型MMA 患儿就诊年龄为1天至9岁10个月, 其中8例来自新生儿筛查(42.1%)。临床表现无特异性,以喂养困难、发育落后等为首发症状,多数于1岁内发病。11例非新生儿筛查的患儿中,首发症状中视力障碍5例(落日眼伴间断下视1例、眼睛下视1例、双眼凝视2例及眼球震颤1例);合并贫血10例;合并肝脏损伤7例;合并肾损伤(尿潜血、尿蛋白阳性)2例;血氨升高5例;乳酸升高3例;血脂升高1例;合并亚临床甲状腺功能减退1例;合并严重骨强度不足1例。见表1。

表1 19例cblC型MMA患儿临床特点
2.2影像学检查 10例行脑电图检查,其中4例脑电图表现异常,2例伴有癫痫发作的患儿呈癫痫波,1例前头部多灶性尖波、尖慢波发放,1例脑电发育轻度迟缓伴前头有一过性尖波。11例行心脏超声检查,其中有心脏异常表现的5例,合并卵圆孔未闭2例,合并肺动脉高压1例,合并少量的三尖瓣反流1例,合并心脏扩大1例。17例行头颅MRI检查,其中表现异常7例,合并陈旧性脑出血2例,脑积水3例、脑室增宽2例,枕大池扩大2例,后纵裂池密度增高1例,合并髓鞘后延迟3例,脑发育不良1例,蛛网膜囊肿2例,脑外间隙增宽3例,额顶叶脑白质密度减低1例,硬膜下积液2例。见表1。
2.3串联质谱分析及有机酸测定 13例进行Met测定,减少4例(30.8%);17例进行HCY测定,增高16例(94.1%);15例进行C2测定,降低2例(13.3%);16例进行C3测定,升高10例(62.5%);18例进行C3/C2检测,升高14例(77.8%);13例进行C3/C0测定,升高8例(61.5%)。17例进行尿甲基丙二酸测定,升高16例(94.1%);15例进行甲基枸橼酸-4测定,升高7例(46.7%)。见表2。
2.4基因检测 17例进行基因检测,共发现MMACHC基因突变13种:c.609G>A(12例)、c.656_ c.658delAGA(4例)、c.80A>G(4例)、c.658_ 660delAAG(3例)、c.567dupT(2例)、c.440-441 delGT(1例)、c.482G>A(2例)、c.81+1G>A(1例) 、c.566_ c.567insT(1例)、c.394C>T(1例)、c.4G>T(1例)、c.467G>A(1例)、c.626dupT(1例)。其中,最常见的基因型为c.609G>A,其次为c.80A>G、c.656_ c.658delAGA。17例中仅1例为纯合,其他均为杂合。13种基因类型中c.81+1G>A、c.656_ c.658idelAGA、c.566_ c.567insT共3个突变未见报道。见表2。
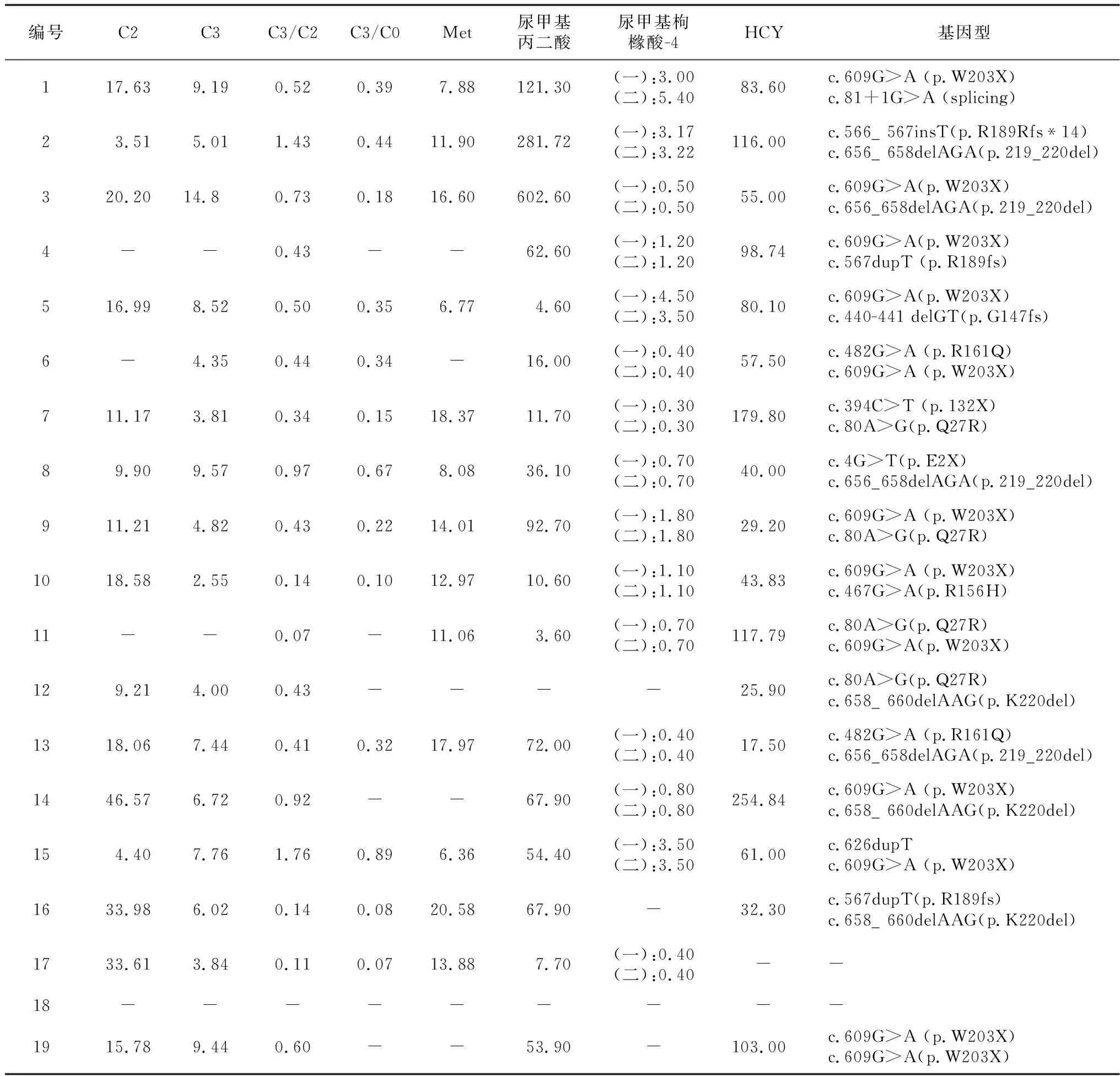
表2 19例cblC型MMA 患儿串联质谱分析、有机酸测定及基因型
3 讨 论
cblC型是我国MMA合并型中最常见的类型,主要以甲基丙二酸蓄积为主要特征,伴有HCY水平的升高。cblC型的突变基因是位于1号染色体上p34编码区的MMACHC基因,该基因编码的蛋白在胞浆内催化还原脱氰反应及合成腺苷钴胺素反应[5]。cblC型患儿相对其他类型发病较晚,临床表现差异大,主要表现为智力落后、巨幼红细胞性贫血、生长障碍等。因此,具有上述表现的患儿不能忽视遗传代谢病的可能性。由于MMA在人群中发病率相对较低,临床表现无特异性,导致该病极易误诊[6]。因此,加强对该病的认知是必不可少的。
MMA患儿体内异常蓄积甲基丙二酸等代谢产物,引起线粒体功能障碍,进而出现神经系统症状[7]。本研究19例中8例通过新生儿筛查发现。11例有首发症状的患儿均以神经系统症状为首发表现,其中8例在首发症状上合并喂养困难、发育落后、反应差,5例首发症状合并视力障碍(落日眼伴间断下视1例、眼睛下视1例、双眼凝视2例及眼球震颤1例),3例伴有抽搐、癫痫发作,2例合并有肺炎。本研究中有3例首发表现合并神经系统症状如癫痫等。17例行头颅MRI检查,其中有7例表现异常,但均无特异性,其中最常见的表现为脑积水、脑室增宽及脑外间隙增宽。因此,cblC 型MMA患儿无论有无神经系统症状,都应定期进行头颅MRI等检查,早期发现神经系统病变,防止出现抽搐、脑积水等严重并发症。对于有发育落后、癫痫等神经系统症状的患者也应做遗传代谢筛查以排除MMA的可能。本研究19例中贫血10例,合并肝脏损伤7例,合并肾损伤2例,乳酸升高3例,血脂升高1例,合并亚临床甲状腺功能减退1例,合并严重骨强度不足1例,合并心脏异常5例,脑电图异常4例,合并头颅MRI检查异常7例,合并高氨血症5例。单纯型更容易表现出高氨血症,本研究中患儿血氨异常表现的相对较少,符合既往相关报道[8]。多系统检查异常表明该病可合并多脏器损伤,建议MMA患儿进行多系统检查,以防止其他系统疾病进展严重。
本研究中,串联质谱检查发现C3/C2、HCY、 尿甲基丙二酸增高人数多于其他检测指标,C0、C2、C3、Met等指标变化人数较少。本研究中全部患儿血HCY升高,与合并型的分型特点保持一致。不同地区常见突变位点不同,我国cblC型MMA患儿中以 c. 609G>A和c. 658 _ 660 delAAG突变较多,两者均发病较早,症状较重,预后较差[9],与本研究结果相一致。本研究中有17例进行基因检测,发现共包括c.609G>A、c.567dupT、 c.482G>A、c.394C>T、c.80A>G、c.4G>T、c.467G>A、c.626dupT、c.566_ c.567insT、c.656_ c.658delAGA、c.658_ 660delAAG、c.440-441 delGT、 c.81+1G>A在内的13种突变类型,其中以 c.609G>A突变位点最常见,其次为c.80A>G、c.656_ c.658delAGA。在我国早发型MMA(发病年龄≤1岁)较晚发型(发病年龄≥1岁)多见[10]。常见的其他早发型基因包括c.440_441del、c.567dupT、c.656_ c.658delAGA等,且携带早发型基因患儿预后相对较差;常见晚发型基因有c.394C>T、c.482G>A、c.615C>A等[11]。欧美国家以c.271dupA突变最多见,中东地区的迟发型患儿以c.394C >T较多见[12]。c.482G>A为常见的晚发型基因突变位点,早期患儿多通过新生儿筛查发现,发病较晚,多以神经系统症状为主并伴有其他多脏器损害,症状相对较轻,早期发现,早期治疗,预后相对较好[13]。本研究中共有2例携带c.482G>A突变位点,均为新生儿筛查发现。由于晚发型MMA特点为症状晚发,早期往往容易忽略,最终影响预后,因此新生儿筛查十分必要。
脑积水是由于脑脊液的产生和吸收不平衡,导致脑室扩大、颅内压升高,严重者导致脑损伤。有研究对9 117例高危儿童进行筛查,结果显示147例MMA患儿中合并脑积水10例,MMACHC基因发生复合杂合突变9例,其中以c.609G>A突变最为常见;MUT基因发生突变1例[14-15]。本研究中3例脑积水患儿基因突变位点包括c.609G>A、c.440-441 delGT、c.567dupT、c.658_ 660delAAG、c.656_ c.658delAGA,多为终止突变或移码突变,对蛋白结构影响较大,从而导致致病性较强,疾病多早发且临床表现严重[16]。当cblC型患儿合并脑积水等严重并发症时,药物控制治疗效果不佳,多采取手术治疗。由此可见突变位点在一定程度上可影响临床表现,对于cblC型患儿应注意预防脑积水等严重神经系统并发症,以提高患儿的生存质量和生存率。
随着技术的不断提高,检测手段也在不断增多。最新研究显示可通过PCR-高分辨率熔融曲线法(PCR-HRM)快速检测MMACHC基因热点致病变异c.609G>A[17]。该病越来越早地被发现和诊断,甚至不少无症状患者在临床症状出现前已得到及时治疗,因此新生儿筛查对于早期发现十分有必要。但在全部检查中,基因检测依然是确诊及分型最可靠的方法。有相应可疑家族史的患儿也应警惕该病,并及时检查、早期诊断。
cblC型MMA的预后主要取决于分型、发病年龄、诊治年龄和是否得到规律治疗[18],起病越早相对症状越重、预后越差。本研究结果表明,1岁内发病、实验室检查数值相对较高10例,症状表现较1岁后发病患儿重。即使早期没有临床表现也不能掉以轻心,部分患儿在没有代谢疾病的情况下可能发生隐性发育延迟,或者在先前健康的儿童、青春期或成人中出现神经功能恶化[19]。本研究中新生儿检出率较高,证明随着生活水平的提高,人们对于新生儿筛查的重视度不断提高,越来越多的新生儿疾病在早期得到发现并及时治愈。我国多采取饮食限制,给予维生素B12、叶酸、左旋肉碱、甜菜碱等药物方法治疗,部分并发症严重患儿,如并发重度脑积水患儿可采取脑积水脑室腹腔分流术等手术治疗。尽管常规治疗可改善生化指标,但cblC型MMA患儿的长期预后并不令人满意,部分早发型病情重、进展快、好转慢,多遗留后遗症[20]。本研究通过对19例患儿的临床资料进行总结分析,对该病的诊断提供了一定的帮助。希望未来有更多准确、早期的诊断方式出现,以减少该病的死亡率及致残率,提高患儿生活质量,延长寿命。
猜你喜欢
分子催化(2022年1期)2022-11-02
健康必读·下旬刊(2020年5期)2020-05-29
特别健康·下半月(2020年4期)2020-04-09
保健与生活(2020年3期)2020-03-02
佛山陶瓷(2018年6期)2018-09-14
中老年健康(2016年10期)2016-11-19
米娜·女性大世界(2016年8期)2016-08-17
Coco薇(2016年5期)2016-06-03
爆笑show(2015年9期)2015-10-24
中学理科·综合版(2008年11期)2008-01-14
