
IGF1-PI3K-induced physiological cardiac hypertrophy:Implications for new heart failure therapies,biomarkers,and predicting cardiotoxicity
2022-01-20 07:00SebstinBssStringerCelesteTiJulieMcMullen
Sebstin Bss-Stringer,Celeste M.K.Ti,Julie R.McMullen,c,d,*
a Baker Heart and Diabetes Institute,Melbourne,VIC 3004,Australia
b Department of Physiology,Anatomy and Microbiology,La Trobe University,Bundoora,VIC 3086,Australia
c Department of Diabetes,Central Clinical School,Monash University,Melbourne,VIC 3004,Australia
d Department of Physiology and Department of Medicine Alfred Hospital,Monash University,Melbourne,VIC 3004,Australia
Abstract
Keywords: Cardiac protection;Cardiotoxicity;Exercise;Heart failure;IGF1;PI3K;Therapies
1. Introduction and background
In this review, we have focused on a signaling cascade in the heart referred to as the insulin-like growth factor 1(IGF1)-phosphoinositide 3-kinase (PI3K) pathway, which plays an essential role in mediating the protective actions of regular physical activity or exercise on the heart.Regular exercise is a well-established and accessible intervention that has been demonstrated to provide benefit to multiple organ systems in settings of both health and disease.1The benefits are well-established in a setting of cardiac health, in which exercise has been demonstrated to reduce the risk of future cardiac events or diseases and improve outcomes following a cardiac event or diagnosis.2,3This is of particular interest given the rising prevalence of ischemic heart disease,which is currently the greatest burden of disease globally, both reducing quality of life and increasing overall mortality.4
Heart failure represents the end point of a variety of cardiovascular diseases and occurs when the heart is unable to supply adequate blood to the body. It is of particular relevance because of the high mortality rate (5-year mortality greater than 40% following initial diagnosis), high lifetime risk of acquisition (20%-45%),5and the limited effectiveness of treatment options currently available. Aerobic exercise training has proven to be one of the few safe and effective interventions following a diagnosis of stable heart failure, with patients displaying improved cardiac function,aerobic capacity, and attenuation of abnormal cardiac remodeling following 3-6-month training programs.6,7Both a lack of patient adherence and an inability to exercise due to loss of cardiac function from heart failure progression pose barriers for an individual to use exercise training as a method of treatment.
Understanding the key molecular pathways and mediators involved in exercise-induced heart protection is an exciting approach for treating heart failure. That being said, the development of an exercise-based therapy is far from a simple process.The cardioprotective effects of exercise reflect a complex and multifactorial web of neurohormonal, hemodynamic,molecular, and physiological changes that occur during and following physical activity, both in settings of acute and chronic exercise.1Exercise in an acute setting activates the sympathetic nervous system and reduces parasympathetic activity. This, in conjunction with engagement of muscular and respiratory pumps,increases stroke volume and heart rate,which in turn leads to greater cardiac output to compensate for an increased demand for oxygen. Chronic or long-term exercise similarly leads to increased sympathetic activity,but additionally, the stimulation of various hormones and growth factors that facilitate the thickening and enlargement of the heart.1
2. Delineating key molecular pathways by understanding differences between the athlete’s heart and the diseased heart
The athlete’s heart is a term coined as far back as 1896 when Henschen8observed cross-country skiers to have enlarged hearts.More recently,this phenomenon has been routinely observed in endurance athletes,who display an increase in heart mass while maintaining preserved or enhanced systolic and diastolic function.9-11Exercise-induced heart growth,also known as physiological cardiac hypertrophy, is a compensatory mechanism that allows for the preservation or enhancement of cardiac function while facilitating the demand for greater cardiac output (increased workload).1This type of growth is typically characterized by an increase in cardiomyocyte size, left ventricular chamber size, wall thickness, and mass. These adaptions function to normalize wall stress and tension in a coordinated manner.12,13
In contrast,pathological heart growth can occur in a setting of disease(pressure overload,myocardial infarction(MI),and cardiomyopathy)and can initially be characterized by thickening of ventricular walls and increased mass,but over time this can lead to cell death, fibrotic replacement, impaired cardiac function,and increased risk of heart failure.12,13Of note,it has been documented that extreme amounts of high-intensity endurance exercise can lead to an increased risk of arrhythmia and/or sudden cardiac death.14The risks from these extreme levels of exercise are distinct from the beneficial, normal levels of exercise that are discussed within this review. In a setting of moderate exercise,an increased risk of arrhythmia and sudden cardiac death is not apparent.
Our laboratory and others1have investigated key mediators responsible for physiological hypertrophy by studying molecular changes in mouse models following chronic exercise (e.g.,swim training)or genetic mouse models.Mice have proven to be a powerful tool to assess key mechanisms responsible for exercise-induced hypertrophy and protection because genes can be relatively easily manipulated to generate knock out and transgenic models,and mice develop significant physiological cardiac hypertrophy after as little as 3-4 weeks with swim training. Moreover, they breed rapidly, are inexpensive to house and functional changes can be assessed through a variety of exercise models(swim,treadmill,and voluntary running).15Numerous molecular pathways have been shown to directly contribute or associate with aspects of physiological cardiac hypertrophy and protection. A comprehensive list is detailed in Bernardo et al.1and includes IGF1-PI3K signaling,mediators downstream of vascular endothelial growth factor,hepatocyte growth factor, and platelet-derived growth factor,neuregulin 1,transcription factors and microRNAs(miRNAs).We have summarized work related to the IGF1-PI3K pathway and strategies for targeting this pathway in the failing heart.
3. The IGF1-PI3K signaling pathway:A key mediator of physiological hypertrophy and cardioprotection
Activation of the IGF1-PI3K pathway through physical activity has been well-established in playing an important and beneficial role in protecting the heart.However,exercise is an activity involving the whole body,and evidence has indicated that exercise also plays an important role in activation of the IGF1-PI3K pathway in both brain and skeletal muscle;14,16-20the impact of exercise on this pathway in other tissue types is less clear.
In this review,we have focused on the IGF1-PI3K signaling pathway because, to date, this pathway is the most recognized and essential signaling pathway responsible for mediating physiological hypertrophy.Cardiac IGF1 formation has been demonstrated to be elevated in elite athletes (soccer players) with enlarged hearts following exercise training. It is postulated that, in response to increased stroke volume during exercise, cardiac myocytes undergo stretch, and IGF1 is released from cardiac myocytes in preference to other growth factors (angiotensin II and endothelin-1) which are released from myocytes in response to a chronic pathological stimuli.21IGF1 binds to the IGF1 receptor(IGF1R),a plasma membrane receptor from the family of tyrosine kinases, and initiates the activation of 2 well-established, pro-hypertrophic canonical signaling pathways—the PI3K-protein kinase B (Akt) pathway and the extracellular signaling kinase pathway.22,23Interestingly, in the adult mammalian heart, PI3K rather than extracellular signaling kinase,is the critical regulator of physiological heart growth.24In this review, we primarily focus on the IGF1-PI3K pathway in the heart,with a particular emphasis on PI3K, providing an updated perspective on current knowledge,the development of therapeutic strategies for heart failure,biomarkers,and predictive tools for cardiotoxicity.
3.1. PI3K signaling
PI3Ks are a family of lipid and protein kinases expressed in all tissues and involved in a wide variety of processes such as cell survival, protein synthesis, cell motility, cell polarity,metabolism, and vesicle trafficking.25Three classes of PI3Ks exist (I, II, and III) that function to catalyze the phosphorylation of phosphatidylinositols to generate class dependent phosphoinositide forms with differing functions.Class I PI3Ks have been researched and understood in depth; there are 4 class I PI3Ks (p110α, β, δ, and γ) that can be further divided in subsets Class IA and IB.Class I PI3Ks function primarily to regulate cell growth, survival, proliferation, autophagy, and metabolism. Research into the function of Class II PI3Ks has been less studied, but 3 isoforms exist (PI3KC2α, PI3KC2β,and PI3KC2γ) with increasing evidence suggesting that they have distinct cellular roles, including cell proliferation, survival, and migration. A single Class III PI3K is conserved in eukaryotes, vacuolar protein sorting 34 acts to phosphorylate phosphatidylinositol to produce phosphatidylinositol 3-phosphate, which regulates autophagy and endocytic sorting.26-28Class I PI3Ks are heterodimers consisting of a regulatory subunit and a catalytic subunit. Multiple isoforms or splice variants of each subunit exist which act to serve differing functions in different cell types.29This review focuses on the p110α isoform of PI3K, a Class IA PI3K30that is primarily activated by tyrosine kinase receptors, and expressed in cardiac myocytes to induce physiological myocyte growth. Activation of PI3K requires interaction of the p85 regulatory subunit with the p110α catalytic subunit,and this catalyzes the conversion of phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol (3,4,5)-trisphosphate. Following this conversion to phosphatidylinositol (3,4,5)-trisphosphate, pleckstrin homology domain-containing proteins including Akt and phosphoinositide-dependent kinase-1 are recruited to the plasma membrane. This recruitment leads to phosphoinositide-dependent kinase-1 and mammalian target of rapamycin complex 2 phosphorylating Akt, which in turn allows for its activation and the triggering of subsequent downstream signaling pathways31(Fig.1).
3.2. Role of PI3K in the heart
Initial interest in the role of PI3K in the heart arose from the previous observation that PI3K plays an essential role in regulating wing size in Drosophila.The insulin/IGF receptor/PI3K pathway is highly conserved across species. In Drosophila with overexpression of wildtype PI3K(Dp110),wing size was significantly larger than wing size from control flies. By contrast, expression of a catalytically inactive PI3K mutant(Dp110D954A) in Drosophila wing resulted in reduced wing size.32,33The role of PI3K in the mammalian heart was first discovered through the characterization of transgenic mouse models with increased and decreased cardiac-specific PI3K(p110α) activity. This research demonstrated that PI3K is a critical mediator of physiological postnatal heart growth.Shioi et al.34created a mouse model expressing a cardiac-specific constitutively activated mutant of PI3K(caPI3K),and a mouse model expressing a cardiac-specific dominant negative mutant of PI3K (dnPI3K). The caPI3K transgenic mice displayed increased cardiac PI3K activity, which corresponded to an increase in the size of all chambers and left ventricular (LV)wall thickness, and heart weight to body weight ratio. The dnPI3K transgenic mice had reduced PI3K activity and in turn a reduction in heart weight(Fig.2).Neither of the models displayed any signs of heart failure following a year of observation.
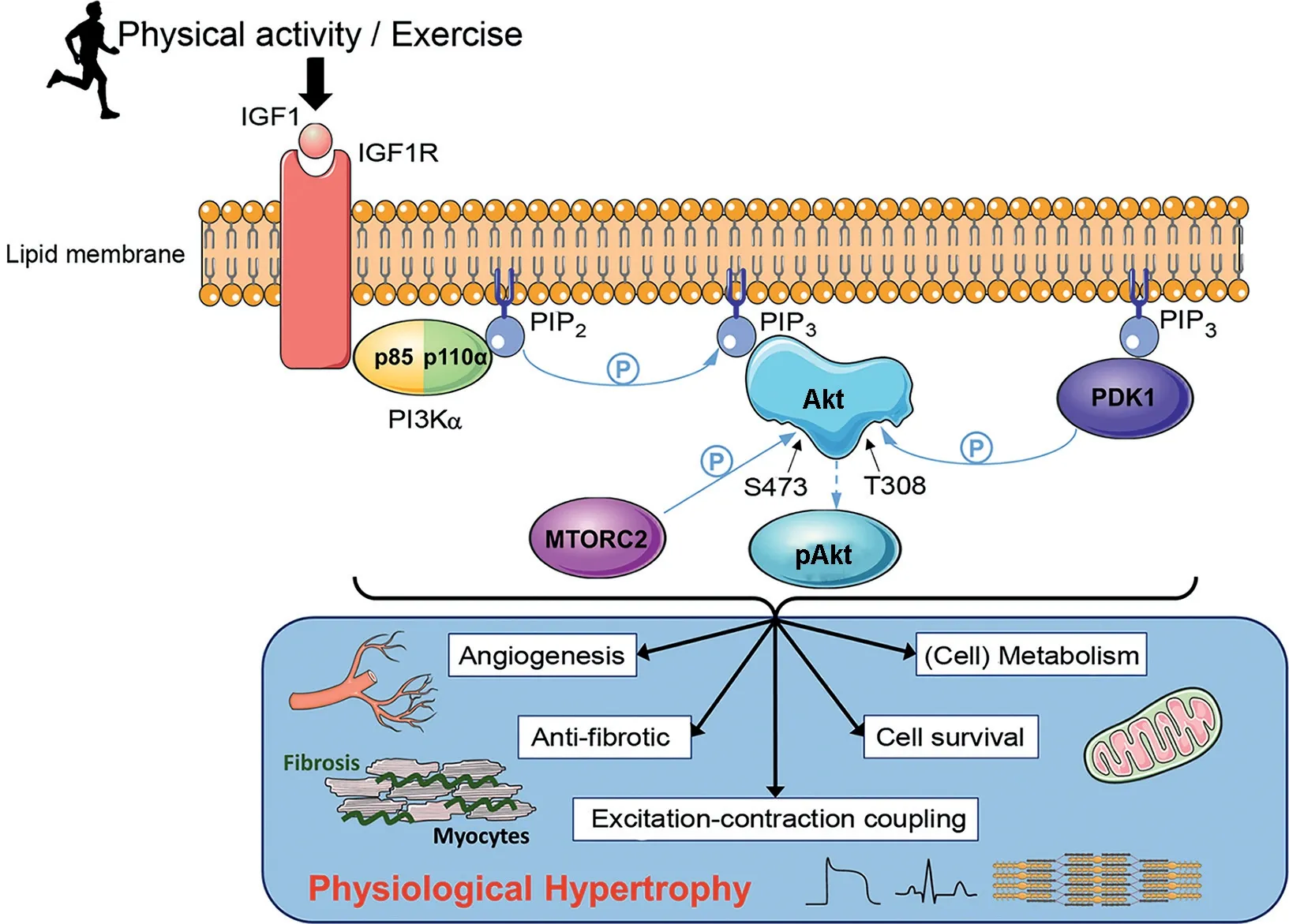
Fig.1. A schematic diagram displaying the impact of physical activity or exercise on the IGF1-PI3K-Akt signaling pathway and the downstream physiological outcomes in the heart.Following exercise,IGF1 binds to the IGF1R embedded in the plasma membrane of cardiomyocytes,allowing for binding of p85,the regulatory subunit of PI3Kα.Once bound,p85 recruits p110α,the catalytic subunit of PI3Kα,forming the fully activated form of PI3Kα.Activated PI3Kα catalyzes the phosphorylation of PIP2 to PIP3, which recruits AKT and PDK1 to the plasma membrane. Binding of Akt to PIP3 causes a conformational change in Akt,exposing the phosphorylation sites S473 and T308.Phosphorylation of S473 by MTORC2 and T308 by PDK1 activates Akt allowing for numerous downstream protective physiological changes to the heart(via Akt dependent and Akt independent mechanisms).P within the blue circle signifies phosphorylation.Akt=protein kinase B;BTK=Bruton’s tyrosine kinase;HER=human epidermal growth factor receptor;IGF1=insulin-like growth factor 1;IGF1R=insulin-like growth factor receptor;MTORC2=mammalian target of rapamycin complex 2;NRG1=neuregulin 1;PDK1=phosphoinositide-dependent kinase 1;PIP2=phosphatidylinositol 4,5-bisphosphate;PIP3=phosphatidylinositol(3,4,5)-trisphosphate;PI3K=phosphoinositide 3-kinase;S473=serine 473;T308=threonine 308.
The role of PI3K(p110α) for the induction of exerciseinduced physiological hypertrophy was later assessed by subjecting dnPI3K mice to chronic swim training. After 4 weeks of chronic swim training the hypertrophic response (heart weight to body weight ratio)of dnPI3K mice was significantly smaller than that of age- and weight-matched non-transgenic(Ntg)controls.35
In addition to its role in regulating heart growth, PI3K has also been demonstrated to mimic the cardioprotective properties of exercise.Exercise training in a genetic model of dilated cardiomyopathy (DCM) from 4 of weeks age, increased lifespan by ~20% and ~16% in male and female mice, respectively. By genetically crossing the DCM model with either dnPI3K or caPI3K mouse models,the impact of altered cardiac PI3K activity on lifespan was also assessed. DCM-caPI3K(increased PI3K) double transgenic mice (without exercise)showed an increase in longevity that was comparable to the increase in lifespan in the DCM model with exercise. In contrast, DCM-dnPI3K (reduced PI3K) mice displayed a drastic reduction in lifespan, highlighting the significance of both exercise and cardiac PI3K activity in the prevention of cardiac disease.36These findings have been replicated in a variety of settings of cardiac stress,with the caPI3K transgenic mice displaying better cardiac function and less pathology following induction of pressure overload, MI, diabetic cardiomyopathy,and atrial fibrillation. Alternatively, the dnPI3K mice consistently display accelerated heart failure and other pathological complications in the above models of cardiac stress.36-41These studies together highlight(1)the importance of PI3K in facilitating normal cardiac growth,(2)the role of PI3K in exercise-induced heart growth, and (3) the critical role of PI3K activity providing protection in a variety of settings of cardiac stress.
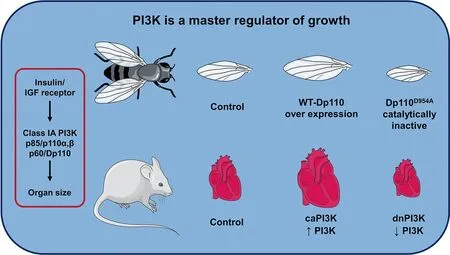
Fig. 2. PI3K is a master regulator of growth. Class IA PI3K(Dp110) overexpression in the wings of drosophila results in enlarged wings while over expression of a mutated inactive PI3K(Dp110D954A)results in smaller wings.Similarly, caPI3K in the hearts of mice results in enlarged hearts, while the presence of a truncated mutated dnPI3K with reduced PI3K activity results in smaller hearts. caPI3K=constitutive activation of PI3K; dnPI3K=dominant negative PI3K; IGF=insulin-like growth factor; PI3K=phosphoinositide 3-kinase;WT=wild type.
In keeping with its crucial role in exercise-induced cardiac growth,PI3K signaling and exercise both target and modulate many of the same cell types and cellular processes.These have been extensively described previously and include the regulation of cardiac myocyte growth, excitation and contraction coupling, vascular adaptions, cellular stress response, mitochondrial adaptations,and anti-fibrotic properties1(Fig.1).
4. PI3K-based therapies as an approach for improving function of the failing heart—Overview
Heart transplantation availability is extremely limited,with as few as 4000-4500 heart transplantations occurring worldwide each year.42Current approaches and strategies under investigation are broadly summarized in Fig.3 and include(1)environmental and dietary approaches, (2) gene-based therapies(e.g.,targeting DNA,mRNA,and miRNAs), (3)pharmacological approaches, and (4) surgical approaches (e.g., LV assist devices, valve replacement, and coronary artery bypass surgery).The majority of existing heart failure treatments primarily manage symptoms and delay disease progression, and exercise interventions are not always viable due to the progressive and debilitating nature of the disease.
Given these cardioprotective benefits that PI3K has been demonstrated to provide a therapy that upregulates cardiac PI3K activity may provide a promising approach for improving heart function in individuals with heart failure.Our laboratory has a large interest in investigating the development of a PI3K based therapy as a non-surgical alternative for the treatment of the failing heart.Multiple approaches may be applicable in considering the use of a PI3K therapy, with differing tactics being tailored to particular pathological or clinical settings (Fig. 3). This is an important consideration given that a single approach is unlikely to be a one fix for all.Factors such as the method of administration(surgical vs.dietary),duration of therapeutic effect (short term vs. long term), and dosage of the PI3K therapy provided may differ depending on an individual’s age, health status, prognosis, and particular cardiac disease being targeted. Moreover, combined therapeutic approaches to treat cardiovascular diseases are more frequently being utilized and are showing encouraging outcomes in recent times.43,44
Approaches our laboratory are investigating include PI3K therapies that utilize adeno-associated virus (AAV)gene therapy, as well as the identification and utilization of mRNAs, miRNAs, small molecules, and lipids that differ between healthy and diseased hearts. Gene therapy is discussed in Section 5, and other approaches are discussed in Section 7.
5. Gene therapy as an approach for treating heart failure
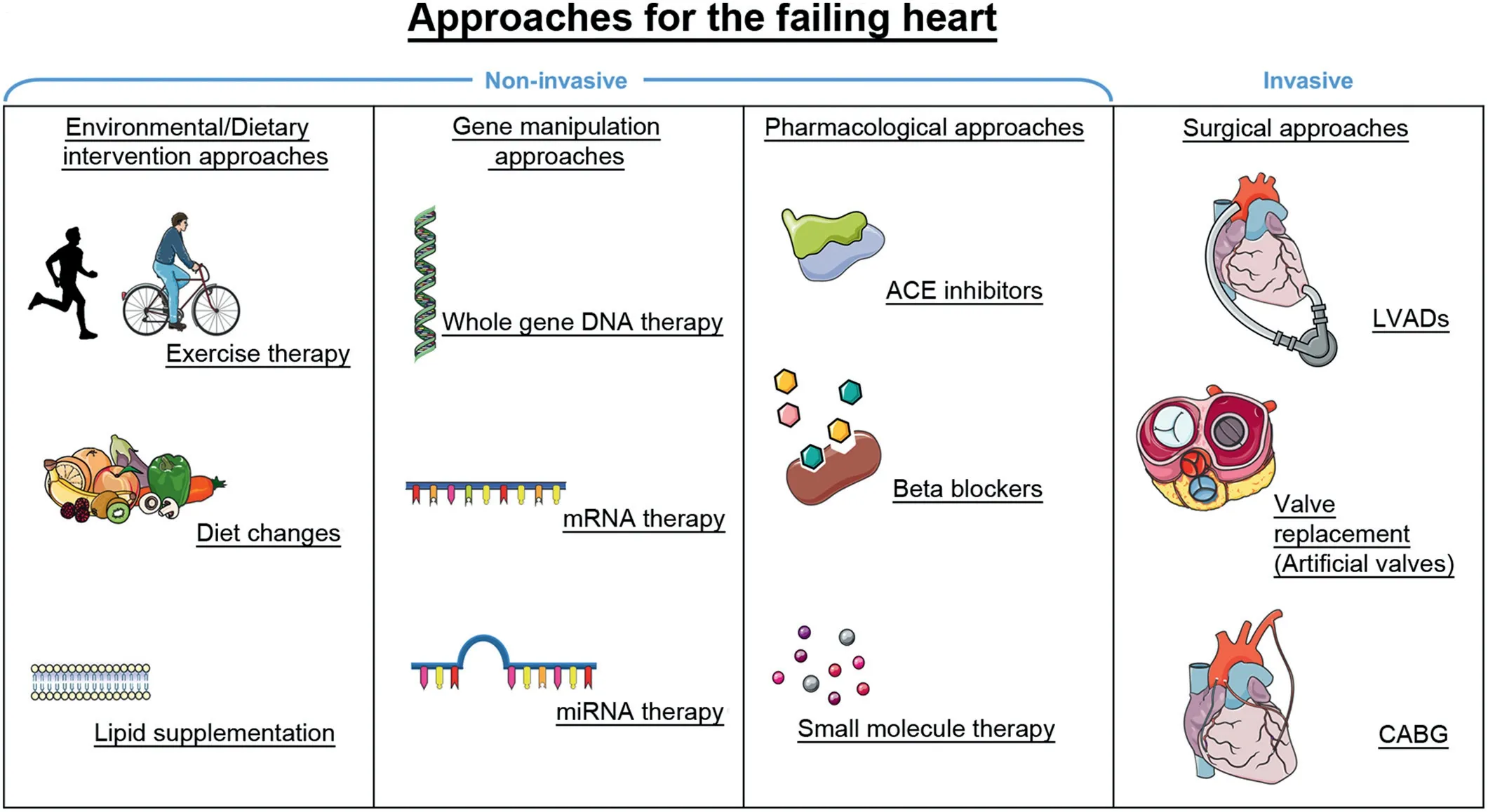
Fig. 3. A summary of current approaches for the failing heart and new promising approaches for improving function in the failing heart (e.g., gene therapy).Approaches are separated into 4 categories based on the type of intervention:environmental and dietary based interventions,approaches involving genetic manipulation,pharmacological interventions,and surgical interventions.Combining multiple approaches may be an optimal strategy to maximize therapeutic outcomes.ACE=angiotensin-converting enzyme;CABG=coronary artery bypass graft;LVAD=left ventricular assist device;miRNA=microRNA.
Gene therapy involves the transfer of an isolated nucleic sequence from a foreign body to a host organism with the purpose of altering gene function and/or expression, and in turn, providing a therapeutic outcome.45Numerous factors need to be taken into consideration to maximize clinical viability when developing a cardiac gene therapy. Namely, the choice of vector used to transfer the transgene, the method of delivering the vector to the heart,the vector’s capacity to provide efficient transduction of the human heart, the therapeutic potential of the transgene of choice, and the cost/practicality of large-scale manufacturing.46
Modified viral vectors have been the primary choice of vectors used for the transfer of genetic material to date;more specifically, AAVs have been demonstrated to be a promising gene therapy vector for the treatment of heart failure with human clinical trials having been undertaken in recent times.47-51AAVs are small, single stranded non-pathogenic viruses with the capacity to transduce both dividing and nondividing cells.The interest in AAVs as a vector for the use in cardiac gene therapy arises from their high safety profile,non-pathogenicity(invoking a limited host immune response),and mediating long-term transgene expression that is reported to last several years in human trials.52Moreover,multiple naturally occurring AAV serotypes such as AAV6 and AAV9 have been found to display cardiac-specific tropism, allowing for efficient transduction of cardiomyocytes while minimizing the delivery of transgenes to non-cardiac tissue or cells.46
Successful results from a multitude of AAV studies to treat heart failure in clinically relevant small and large animal models paved the way for the 1st AAV heart failure trial in human subjects: The Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) trial.The CUPID trial attempted to increase expression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase(SERCA2a)activity,which is reduced in the failing heart. Administration of AAV1-SERCA2a in a pilot study in 9 patients with heart failure displayed a positive safety profile and favorable outcomes including improved ejection fraction,end-systolic volume,and maximum rate of oxygen consumption (Table 1).49This trial was replicated in a follow-up placebo controlled, Phase IIaCUPID trial of 39 patients,in which those that received a high dose of AAV1-SERCA2a (1×1013DNase-resistant particles) demonstrated significantly improved LV end-systolic volume and maximum rate of oxygen consumption,as well as a decreased frequency of cardiovascular events and cardiovascular related hospitalizations (Table 1).50Subsequently, a larger multinational,randomized,double-blinded placebo-controlled CUPID Phase IIb trial with 243 advanced heart failure patients was initiated. The Phase IIb trial did not demonstrate improvement in the primary or secondary end point of recurrent heart failure events or all-cause death, respectively(Table 1).The trial was prematurely terminated,but of importance, no signs of adverse safety outcomes were observed across all studies at any dose following administration of AAV1-SERCA2a.51The outcome of the CUPID Phase IIb trial could not conclusively assess whether AAV1-SERCA2a was an appropriate gene target for the treatment of heart failure because efficiency of transduction was considered suboptimal.However,the results have been invaluable in informing future efforts of the use of AAV gene therapy for the treatment of cardiovascular disease.

Table 1 Outcomes from the AAV-SERCA2a CUPID trials.
The poor outcomes from the CUPID Phase IIb trial have not dampened efforts to utilize AAVs as a gene therapy vector,with over 200 clinical trials involving AAVs being registered at Clinicaltrials.gov as of June 2020,ten of which are categorized under the topic of heart disease. The aforementioned caPI3K transgene represents another gene target for the treatment of heart failure. Preliminary studies using an AAV6-caPI3K with a cytomegalovirus (CMV) promoter in both healthy mice and models of cardiac pathology have demonstrated efficacy regarding its potential as a cardioprotective therapeutic agent.40The AAV6 serotype in conjunction with use of a CMV promoter provided cardiac and skeletal muscle-specific transduction. Moreover, administration of AAV6-caPI3K-induced angiogenesis, physiological hypertrophy,and increased phosphorylation of Akt in the hearts of healthy mice.Similarly,promising results have been observed using AAV6-caPI3K in multiple models of established cardiac pathology. In a mouse model with established cardiac dysfunction due to pressure overload(transverse aortic constriction),AAV6-caPI3K was able to restore systolic function(fractional shortening) within 10 weeks of administration.40AAV6-caPI3K administration also provided cardiac protection in mouse models with type 1 or type 2 diabetes.The type 1 diabetic model (low-dose streptozotocin) displayed diastolic dysfunction prior to AAV6-caPI3K,and this was attenuated within 6-8 weeks post-AAV.The type 2 model of diabetes(low-dose streptozotocin in combination with a high-fat diet)displayed systolic dysfunction prior to treatment, and AAV6-caPI3K increased systolic function within 8 weeks.Both type 1 and 2 diabetic models displayed cardiac fibrosis,and fibrosis was lower in AAV6-caPI3K treated mice compared to the corresponding diabetic control mice.41,53
Collectively, these findings have facilitated the continued optimization of AAV6-caPI3K as a gene therapy tool,and its transition into large animal models, a crucial steppingstone between the laboratory and the clinic.
In the process of the development and translation of a gene therapy such as AAV-caPI3K,from a laboratory to a clinical setting, 3 important considerations must be addressed: first,the cardiac specificity of the therapy (ensuring that the transgene is highly expressed in cardiac tissue while simultaneously not displaying expression in non-cardiac tissue);second, the feasibility of mass AAV production to ensure the therapy is financially viable as a treatment option for patients with heart failure; and third, demonstrating safety and efficacy in a large animal model of heart failure.
5.1. Cardiac specificity
Increased PI3K activity is protective and beneficial in cardiac tissue,but PI3K is well-known to be a regulator of tumor growth in other tissues in a variety of cancers.54,55Cardiac myocytes within the adult heart have very little capacity to proliferate.Thus,changes in heart size and mass are typically a consequence of changes in cardiac myocyte size. In response to PI3K activation, cardiac myocytes enlarge and this results in physiological hypertrophy. However, in many forms of cancer, dysregulation and increased activation of the PI3K/Akt pathway in other cell types can lead to uncontrolled cell proliferation and growth, as seen in settings of tumorigenesis.56Ensuring cardiac-specific gene transfer of a PI3K gene therapy is a crucial safety consideration for the prevention of undesirable side effects such as the development of cancer.
Another important consideration is efficient cardiac transduction.If a viral capsid is unable to efficiently transduce cardiac tissue, its capacity as a therapeutic agent is made redundant. One approach to address these concerns involves the selection of AAV serotypes that are naturally cardiotropic.Multiple studies have compared the effectiveness of the most well-established AAV serotypes (AAV1-9), and AAV6 and AAV9 have frequently demonstrated rapid and robust cardiotropic expression with substantial expression in the hearts relative to other organs.57-61As described earlier, the AAV6-CMV-caPI3K vector displays transduction specific to cardiac and skeletal muscle.40Further improvement to cardiac-specific transduction can be established using a cardiotropic AAV serotype in conjunction with a promoter that confers cardiomyocyte-specific gene expression such as a cardiac troponin T promoter. Prasad et al.62compared transduction of AAV6 vectors harboring either a CMV or cardiac troponin T promoter with a luciferase reporter in a variety of tissue types in mice.Luciferase expression driven by the CMV promoter was comparable in the heart, skeletal muscle, and liver, whereas luciferase expression driven by the cardiac troponin T promoter was nearly 100-fold greater in the heart than all other tissue types assessed.62In addition to promoter and serotype selection,further improvements to cardiac specificity may be made through the development of promoters that specifically target diseased tissue,development of chimeric AAV vectors through DNA shuffling, or the use of directed evolution to modify viral capsid sequences and select for cardiotropic variants.46
5.2. Feasibility of mass AAV production
The viability of mass scale production of AAV is another necessity to be considered in the process of clinical translation.63The high global prevalence of heart failure together with the costs of mass producing larger yields of AAV for preclinical studies in large animals and for clinically relevant therapeutic interventions would come with a considerable price tag. The limited packaging capacity of AAV (~5 kb), makes obtaining sufficient vector yields of larger genes (such as caPI3K) particularly challenging. Reducing the size of gene constructs is an approach to improving yield and in turn reducing cost.Furthermore,other promising approaches to improve large scale production of AAVs have involved modifications to the culture conditions for growing cells with plasmids used for chemical co-transfection of AAVs, as well the investigation of cell lines which display greater transfection efficiency and in turn improved AAV yields.64
5.3. Translation to large animal models
Assessment of efficacy and safety in large animal models is important for any new gene therapy approach.Sheep and pigs have typically been the model of choice after small animal studies.This primarily reflects the anatomical and physiological similarities shared between these large animals and humans, which differs in rodents.Moving from small animals to large animals invokes additional challenges, such as determining the optimal method to administer an AAV,identifying if either toxicity or efficacy vary in different animal models,and the optimal dose required to provide a therapeutic effect.This topic is covered in detail by Bass-Stringer et al.46
6. Cardiac pathology and cardiotoxicity in settings of reduced PI3K
The earlier part of this review has focused on enhancing PI3K activity in the heart to provide protection in settings of cardiac stress. However, a reduction in PI3K in the heart has the converse effect, that is,making the heart more susceptible to cardiac pathology and heart failure. Factors which can lead to reduced or defective PI3K signaling in the heart include physical inactivity, obesity, diabetes, aging, and drugs (e.g.,anti-cancer drugs). The previously mentioned dnPI3K transgenic mouse model has been a valuable tool for understanding the impact of reduced cardiac PI3K activity in a variety of settings of cardiac pathologies. dnPI3K mice displayed cardiac dysfunction in response to pressure overload compared to Ntg controls. This was demonstrated through a significant reduction in fractional shortening,and a marked increase in systolic and diastolic LV dimensions following 1 week of ascendingaortic banding compared to Ntg-banded controls.The animals also displayed an increase in lung weight/body weight ratio,a marker of LV dysfunction.35,36Similarly, in a setting of MI,dnPI3K mice also displayed reduced fractional shortening and increased chamber dimensions.38
PI3K activity has also been shown to affect the progression of heart failure in a setting of DCM. dnPI3K transgenic mice have been crossed with 2 different cardiac-specific transgenic mouse models of DCM to generate double transgenic mice(dnPI3K-DCM). In the first DCM model, due to very high expression of Cre-recombinase,the dnPI3K transgene drastically shortened life span (~50%) in the dnPI3K-DCM compared to DCM transgenic mice.In a second model of DCM,due to overexpression of mammalian sterile 20-like kinase 1 (Mst1), the dnPI3K-DCM transgenic mice displayed an accelerated heart failure phenotype including more cardiac dysfunction, greater atrial enlargement and cardiac fibrosis than DCM (Mst1) transgenic mice,and developed atrial fibrillation.36,37
In a setting of diabetes,dnPI3K mice have also been shown to develop an exaggerated cardiomyopathy phenotype compared to Ntg diabetic mice.39Taken together, these studies demonstrate that a reduction in PI3K leads to accelerated cardiac pathology and heart failure in a variety of settings of pathological stress.
The IGF1-PI3K pathway is considered a master regulator of cancer in a variety of non-cardiac tissue types.56,65Thus,extensive efforts have been devoted to developing agents that act to inhibit or modify components of the IGF1-PI3K pathway, and in turn improve survival rates for cancer patients.However, with improvements in survival from cancer, some patients are developing cardiac complications including heart failure and arrhythmias. Given the widespread pathological outcomes seen in mice with reduced PI3K activity, consideration should be taken for potential adverse effects due to cardiotoxicity that may arise when inhibiting this ubiquitously expressed pathway in an already compromised population.Kinase inhibitors for the treatment of cancer,such as trastuzumab,66have been recognized to contribute to cardiac dysfunction.67A Phase III randomized multicenter trial68combined trastuzumab with anthracyclines and cyclophosphamide to treat human epidermal growth factor receptor 2(HER2)-positive breast cancer patients. Heart failure and cardiac dysfunction was reported in up to 27% of patients receiving the combined therapy. Comparatively, the group that received only anthracyclines and cyclophosphamide had an incident rate of 8%.68A number of mechanisms have been implicated to explain trastuzumab cardiac toxicity but it is noteworthy that this drug also has the potential to inhibit PI3K signaling via HER2(Fig 4).66,69
Modification of other proteins regulated by the IGF1-PI3K pathway are being targeted for the development of novel anticancer drugs. Clusterin has been implicated in the pathogenesis of various cancers,such as prostate cancers,breast cancers,and lung cancer,70,71and has been a target of interest, with multiple recent clinical trials having focused on silencing clusterin with an antisense oligonucleotide (Custirsen) as a therapeutic intervention. We recently reported a potential role of clusterin in physiological cardiac hypertrophy and cardiac protection. Expression of clusterin was increased in hearts of caPI3K mice and decreased in hearts of dnPI3K mice.In addition, we identified increased secretion of clusterin in media from neonatal rat ventricular myocytes stimulated with IGF1.72Given that a link exists between reduced PI3K activity and clusterin expression72(Fig. 4), the possibility for the development of cardiac toxicity following its silencing should be considered.
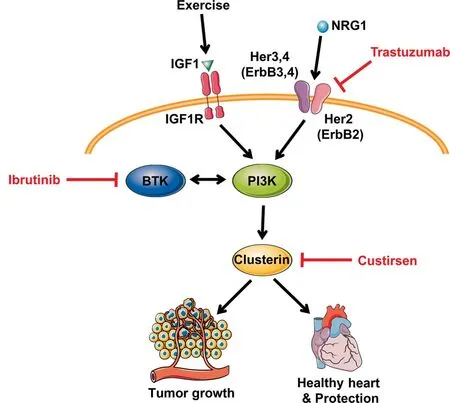
Fig. 4. Anticancer therapeutics with the potential to inhibit the PI3Kα pathway. Trastuzumab binds to the extracellular domain of HER2 and triggers mechanisms to downregulate downstream activity. Ibrutinib inhibits BTK expression and is a known regulator of the PI3K-Akt pathway.Custirsen acts to silence clusterin, of which its expression has been correlated with PI3K activity, and may disrupt downstream processes. The mechanisms of anticancer therapies that suppress tumor growth by targeting the PI3K-Akt pathway may simultaneously contribute to an increased susceptibility for the development of cardiac pathologies. Red lines indicate interventions that act to silence, inhibit, or downregulate protein expression. Akt=protein kinase B;BTK=Bruton’s tyrosine kinase; HER(ErbB)=human epidermal growth factor receptor; IGF1= insulin-like growth factor 1; IGF1R=insulin-like growth factor receptor;NRG1=neuregulin 1;PI3K=phosphoinositide 3-kinase.
Similarly,a Bruton tyrosine kinase inhibitor called ibrutinib is a targeted cancer therapy used for the treatment of many hematological cancers including chronic lymphocytic leukemia, small lymphocytic lymphoma, mantle cell lymphoma,and Waldenstrom macroglobulinemia. Across multiple trials,3.5%-6.5% of subjects receiving ibrutinib treatment developed atrial fibrillation.73There is potential crosstalk between Bruton tyrosine kinase and the PI3K-Akt pathway,74and thus, ibrutinib has the potential to interfere with cardioprotection(Fig.4).Our laboratory has shown that mice with reduced PI3K activity display greater susceptibility to atrial fibrillation and that PI3K-Akt activity is reduced in human atrial appendages from patients with atrial fibrillation.37Moreover,reduced PI3K-Akt expression has been observed following the exposure of neonatal rat ventricular myocytes to ibrutinib.73These observations highlight the importance of taking caution when considering any intervention that may suppress the PI3K-Akt pathway in the heart.The impact of exercise on the IGF1-PI3K signaling pathway in non-cardiac tissue should also be considered when assessing potential toxicities of therapies that act to alter the expression or function of the IGF1-PI3K pathway, as exercise represents a systemic intervention,and has been demonstrated to play a role in activating the IGF1-PI3K pathway in brain and skeletal muscle tissue.16-20Crosstalk between skeletal muscle and other tissues with the heart has also emerged.1
7. Identifying molecular distinctions in the healthy and diseased heart—New drug targets and biomarkers
The physiological and pathological hypertrophic heart display distinct and differing functional, metabolic, structural,and molecular features.The use of surgical,genetic,and exercise models representing pathological or physiological cardiac remodeling, together with the profiling of genes, proteins,lipids,and metabolites,has become a valuable research tool to identify potential new drug targets and biomarkers which are distinct in the healthy and diseased heart (Fig. 5). A detailed overview of profiling studies has been summarized previously.1This same approach can be used in genetic mouse models which are protected or more susceptible to cardiac stress(e.g., caPI3K and dnPI3K transgenic mice, respectively). Distinguishing a diseased or stress susceptible heart may provide opportunities to assess whether someone is more likely to develop more severe cardiac pathology in response to cardiac stress(e.g.,hypertension)or a cancer therapy.
Our laboratory has undertaken gene-profiling studies to attempt to identify candidate therapeutic genes regulated by PI3K.Assessing the profiles of caPI3K,dnPI3K,and Ntg mice subjected to cardiac stress through MI was used to generate a list of genes that were differentially expressed based on PI3K activity.38Correlating these differentially expressed genes with cardiac function(fractional shortening percentage)and in turn identifying those that are selectively expressed in the heart,provided a shortlist of candidate therapeutic targets.One of the top candidate genes was Acadm.
The protein product of Acadm is medium chain acyl-coenzyme A dehydrogenase(MCAD),a protein that has not previously been linked with physiological hypertrophy and protection. The therapeutic potential of MCAD in the heart was examined by administering an AAV6 vector encoding MCAD to both healthy mice and mouse models of cardiac dysfunction.75MCAD induced physiological hypertrophy in healthy mice and mitigated characteristics of cardiac remodeling in a setting of cardiac pathology due to pressure overload.75This highlights the potential of how assessing differences in the healthy and diseased heart can identify candidates for novel therapies.
The same approach that led to the identification of Acadm was applied to identify miRNAs that are differentially regulated in caPI3K mice,dnPI3K mice,and Ntg mice.Numerous candidates were identified,38and subsequently, silencing or inhibition of miR-34, miR-652, or miR-154 provided benefit when targeted in settings of cardiac pathology(MI and/or pressure overload).76-78
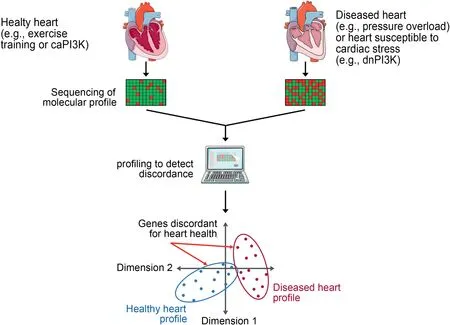
Fig.5. Identifying molecular distinctions between the healthy and diseased heart.A simplified pipeline demonstrating the process of using sequencing technologies to identify candidate therapeutics and biomarkers of cardiac health and disease.Tissue is collected,pooled,and processed from healthy and diseased hearts.A molecule of interest(e.g.,DNA,RNA,protein,lipid)is sequenced and profiled.Expression is compared between groups to identify specific candidates that are discordant for cardiac health,or techniques such as principal component analysis can be used to identify global changes between groups.caPI3K=constitutive activation of PI3K;dnPI3K=dominant negative PI3K;PI3K=phosphoinositide 3-kinase.
Our laboratory has also undertaken comprehensive profiling of the lipidome in models of physiological and pathological remodeling. As cardiac myocytes enlarge or change shape in response to a stimulus(e.g.,cardiac stress such as hypertension or chronic exercise training), the plasma membrane which includes hundreds of lipid species, undergoes dramatic remodeling. Lipid profiling (>300 lipid species) demonstrated that lipid profiles differ substantially in models of physiological cardiac remodeling (swim training, caPI3K transgenic mice, and IGF1R transgenic mice), models of pathological remodeling (severe pressure overload due to transverse aortic constriction, a transgenic model of DCM,and mice with reduced cardiac PI3K activity and greater susceptibility to cardiac stress, i.e., dnPI3K transgenic).79,80As an example,many sphingolipid species were decreased in the hearts of caPI3K mice and increased in the hearts of dnPI3K mice; by contrast, many phospholipids were increased in the hearts of caPI3K mice but decreased in dnPI3K mice.79Dietary supplementation of lipid species that are depressed in the diseased heart and increased in the healthy heart may offer a non-invasive therapeutic approach for improving heart function(Fig.3).
Microarray gene profiling and/or protein analyses in physiological models (IGF1R transgenic mice, PI3K transgenic mice, and exercise-trained mice) also led to the observation that heat shop protein 70 (Hsp70) expression is elevated in the heart with cardiac IGF1R-PI3K signaling and exercise training.24,40Hsp70 plays a key role in the cellular stress response.1Based on this work, we assessed the therapeutic potential of a small molecule which was a known co-inducer of Hsp70 in a mouse model with heart failure and atrial fibrillation. The small molecule (BGP-15) improved heart function, reduced arrhythmia and was associated with lower cardiac fibrosis.Unexpectedly,the small molecule appeared to provide benefit via phosphorylation of IGF1R, which was independent of Hsp70.81
8. Concluding remarks
The benefits of regular physical activity are well-known and represent an accessible intervention that can improve cardiac function and reverse cardiac remodeling in a setting of heart failure.However,patient adherence to exercise is a significant issue. Thus, alternative strategies for recapitulating some of the key benefits of exercise on the heart are of substantial interest. Characterizing mouse models with altered cardiac PI3K activity under basal and disease settings have provided an invaluable tool to identify molecular distinctions between the healthy and diseased heart because PI3K is a critical regulator of physiological cardiac hypertrophy but not pathological hypertrophy. This has allowed for the discovery of novel targets for the treatment of heart failure.Furthermore, the importance of PI3K activity for maintaining cardiac function should be taken into consideration when evaluating the viability of therapies that act to reduce activity of the IGF1-PI3K pathway, as these may cause cardiotoxicity in more vulnerable patients with other conditions such as diabetes and hypertension.
Acknowledgments
All authors are supported by the Victorian Government’s Operational Infrastructure Support Program.SBS is supported by a joint Baker Heart and Diabetes Institute-La Trobe University doctoral scholarship. JRM is supported by a National Health and Medical Research Council Senior Research Fellowship(Grant No.1078985).
Authors’contributions
SBS and JRM drafted the manuscript; CMKT contributed to editing the paper. All authors contributed to the generation of figures. All authors have read and approved the final version of the manuscript, and agree with the order of presentation of the authors.
Competing interests
The authors declare that they have no competing interests.
 Journal of Sport and Health Science2021年6期
Journal of Sport and Health Science2021年6期
- Journal of Sport and Health Science的其它文章
- The association of grip strength with cardiovascular diseases and all-cause mortality in people with hypertension:Findings from the Prospective Urban Rural Epidemiology China Study
- Cardiorespiratory fitness measured with cardiopulmonary exercise testing and mortality in patients with cardiovascular disease:A systematic review and meta-analysis
- The relationships between step count and all-cause mortality and cardiovascular events:A dose-response meta-analysis
- The epigenetic landscape of exercise in cardiac health and disease
- Exploring the impact of COVID-19 on the movement behaviors of children and youth:A scoping review of evidence after the first year
- No independent associations between physical activity and clinical outcomes among hospitalized patients with moderate to severe COVID-19