
一种同时培养原代神经元和原代星形胶质细胞的实验方法
2022-01-19 08:31薛建锋
临床与实验病理学杂志 2021年12期
薛建锋,魏 娜,杨 淳
原代细胞是指从活体获取组织后的首代培养细胞,因其性质最符合体内的状态,因此在药理药效学、毒理学、细胞分化、细胞凋亡、信号转导通路等研究中价值最大。神经元是神经系统主要发挥功能的细胞,而星形胶质细胞是神经胶质细胞中功能最复杂的细胞,在神经系统疾病的研究中常常需要同时对这两种细胞功能状态进行探讨。常规方法是分别对这两种细胞进行提取培养,过程繁琐,且培养细胞的纯度有限。如果能将两者同时提取培养,既可以节约时间、成本,又可满足实验目的需要。因此,本科室利用同一批实验动物同时进行原代神经元及原代星形胶质细胞培养,并对分离培养过程进行优化,以达到理想的实验结果。
1 材料与方法
1.1 试剂和仪器
1.1.1试剂 Neurobasal-A培养基、DMEM/F-12(1 ∶1)培养基、B27(50×)营养因子(Gibco公司);胎牛血清、PBS液(BI);左旋多聚赖氨酸(PLL,Sigma公司);木瓜蛋白酶、D-hanks液、TritonX-100、DAPI、抗荧光淬灭封片剂(索莱宝公司);4%多聚甲醛(Biosharp公司);免疫荧光抗体封闭剂(碧云天公司)、鼠源神经核抗原(NeuN)抗体、FITC结合山羊抗兔抗体、兔源GFAP抗体、山羊抗兔IgG-Alexa Fluor 488(武汉Proteintech公司);兔源MAP2抗体(CST)、Cy3结合山羊抗鼠抗体(Absin公司)。
1.1.2仪器 CO2细胞培养箱、倒置显微镜、细胞计数仪、超净工作台、台式放大镜。
1.1.3主要溶剂的配制 (1)0.1 mg/mL左旋多聚赖氨酸(PLL)溶液:称取PLL后超纯水溶解,配制成1 mg/mL的母液放置于-20 ℃。包被时用超纯水稀释10倍,0.22 μm的滤器过滤备用。(2)2 mg/mL木瓜蛋白酶溶液:称取50 mg木瓜蛋白酶粉末溶于25 mL DMEM/F-12培养基中,现配现用。(3) 神经元培养基:Neurobasal-A培养基、B27、双抗比例为97 ∶2 ∶1。(4)20%DMEM/F-12培养基、10%DMEM/F-12培养基分别为含有20%和10%胎牛血清、双抗1%的培养基。
1.2 实验动物清洁新生24 h内的SD乳鼠(雌雄不限),由贵州医科大学动物房提供,合格证号[SYXK(黔)2018-0001]。
1.3 方法
1.3.1培养皿和培养瓶的预处理 取6、12、96孔板、T25培养瓶进行预处理。其中12孔板预先放置细胞爬片。0.1 mg/mL PLL处理各培养皿,在37 ℃培养箱中包被过夜。第2天用无菌PBS液清洗2次,吸干水分,放入超净台晾干备用。
1.3.2海马神经元取材和培养 (1)取出生24 h内的SD乳鼠,75%乙醇消毒3 min,断脊处死。剪开颈部皮肤,沿失状线将皮肤剪开至鼻端。弯头眼科镊轻轻划开矢状缝下筋膜,然后用两把弯头眼科镊沿枕骨大孔处插入,对称撕开颅骨,暴露整个大脑皮层。将皮层放入预冷的D-hanks液中,在放大镜下用眼科镊沿着枕叶缓慢游离出“新月形”的海马。将海马体的血管剥离干净后放入预冷的种植培养基中。(2)手术刀将海马切碎,大小1 mm×1 mm×1 mm,静置2 min弃上清液,37 °C加入2 mg/mL木瓜蛋白酶消化20 min,每5 min摇晃培养皿,使其充分消化。在显微镜下观察组织块上无细胞后,加入等体积胎牛血清终止消化。向培养皿中加入3 mL种植培养基,用加样枪吹打50次,静置1 min,吸取上清液至离心管,重复此步骤3次,弃掉剩余沉淀。将上清液放入离心机850 r/min离心5 min。弃上层液,加神经元培养基10 mL,200目筛网过滤,制成单细胞悬液。(3)吸取10 μL加入特定的计数板,用BIO-RAD TC20自动细胞计数仪计数,调整种植密度为1.5×106/mL,种植入6、12、96孔板,标记提取日期和细胞种类。培养4 h后,倒掉神经元培养基,用不含血清的DMEM/F-12清洗1遍,加新的神经元培养基,后每隔2天半量换液。(4)分别在24、48、72 h观察细胞形态,拍照记录。第5天用免疫荧光法鉴定12孔板细胞。第7天开始进行后续实验。
1.3.3星形胶质细胞取材和培养 (1)将剩余大脑皮层放入预冷的D-hanks液中,去掉小脑、脑干,更换D-hanks液。直头镊和弯头镊配合,在冰袋上将皮层血管剔除干净。D-hanks液漂洗1次并吸干液体。手术刀将其切为1 mm×1 mm×1 mm大小小块,加入0.25%胰酶,放入37 ℃培养箱消化20 min,期间每隔5 min摇晃1次。巴氏管吸取5 mL 20%DMEM/F-12培养基终止消化,轻柔地吹打60次分离细胞。(2)用200目筛网过滤,去掉大块组织。再加20%DMEM/F-12培养基,放入培养箱差速贴壁20 min。自动细胞计数仪计数,调整种植密度为1×106/mL,种植进相应的培养瓶。(3)梯度血清法纯化:20%DMEM/F-12培养基中培养3天,PBS洗2遍,加入10%DMEM/F-12培养基培养2天。第5天,加不含血清的DMEM/F-12培养基。第7天,用十字手摇法摇晃培养瓶5 min,PBS洗2遍,加10%DMEM/F-12培养基培养,以后每2天换液,均以10%DMEM/F-12培养基培养。选第10天细胞进行鉴定。当细胞长至培养瓶90%时传代培养,0.125%胰酶消化,1 000 r/min离心5 min。选5代以内的细胞进行后续实验。
1.3.4神经元和星形胶质细胞的鉴定 (1)将12孔培养板中的细胞爬片用PBS浸洗2次,每次3 min。(2)用4%多聚甲醛固定爬片20 min,PBS浸洗玻片3次×3 min。(3)0.25%TritonX-100(PBS配制)室温通透20 min。(4)PBS浸洗玻片3次×3 min,用免疫抗体封闭液封闭1 h。(5)吸掉封闭液,在提取海马神经元的爬片上分别滴加鼠源NeuN抗体(1 ∶100)、兔源MAP2抗体(1 ∶100),在提取大脑皮层的细胞爬片上滴加兔源GFAP抗体(1 ∶100),4 °C孵育过夜。(6)第2天,PBST浸洗爬片3次×3 min,在海马神经元的爬片中分别滴加抗小鼠的Cy3结合荧光二抗和山羊抗兔IgG-Alexa Fluor 488,在大脑皮层的爬片上滴加抗兔的FITC结合荧光二抗。暗盒中室温孵育2 h,PBST浸洗3次×3 min,所有操作在暗处进行。(7)滴加DAPI避光孵育5 min,对标本进行染核,PBST浸洗3次×3 min。(8)吸干爬片上的液体,用两把镊子将爬片夹出,置于事先滴加抗荧光淬灭封片剂的载玻片上,将长有细胞的一面朝下,荧光显微镜下观察并拍照。
1.3.5神经元和星形胶质细胞纯度的计算 随机取3个200倍视野,通过对每个视野内NeuN和GFAP阳性细胞数占细胞总数的比例进行分类统计,求平均值。
2 结果
2.1 原代神经元的形态接种12 h后,细胞体积小,呈圆形、椭圆形、不规则形,周边环绕光晕,少数细胞有细小短突起,多数细胞已贴壁生长。换维持培养基1天后,细胞部分呈聚集性增长,胞体增大,突起变长,细胞间突起有少量连接。培养第5、6天的神经元体积变大、成熟饱满,细胞质较丰富,光晕更为明显,神经元突起连接密切。培养第7天,细胞明显聚集,突起增多伸长,细胞之间相互连接形成致密细胞网(图1)。
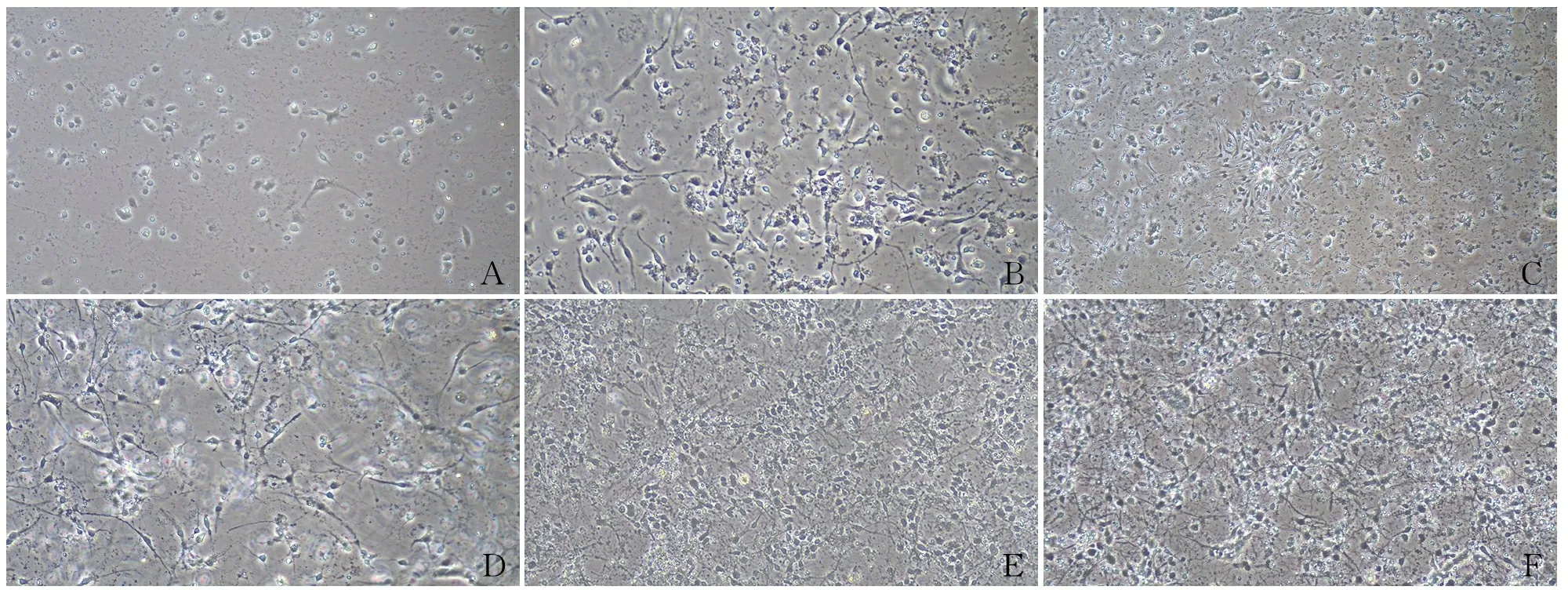
图1 不同时间段海马神经元的形态变化:A. 24 h;B. 48 h;C.72 h;D. 第5天;E. 第6天;F. 第7天
2.2 星形胶质细胞的形态培养第1天,细胞背景较杂,有各种形态:梭形、圆形、不规则形,多数细胞贴壁生长,含有较多的组织碎片;第3天,细胞胞体增大,呈锥形,有数个伸长的突起,里面混有折光性强的小胶质细胞,也有少量多突起的神经元;第5天,细胞胞体进一步增大,突起也较大,相互连接,融合度达70%;第7天,大量细胞死亡,换液后可见剩余贴壁生长的细胞形态较为一致,细胞胞体大,有数个大的突起相互连接,同时含有少量的小胶质细胞(图2)。经第1次传代培养后,可观察到细胞形状呈不规则形,胞质丰富,突触较长且交错分布,呈典型的“石子路”样,为星形胶质细胞。

图2 不同时间段星形胶质细胞的形态变化:A. 第3天;B. 第5天;C. 第7天
2.3 神经元和星形胶质细胞的鉴定分别用细胞特异性抗体NeuN和GFAP鉴定神经元和星形胶质细胞,选取3个视野求得神经元和星形胶质细胞的纯度分别为(95±0.62)%、(94±0.73)%,达到了进行后续实验的纯度要求(图3)。
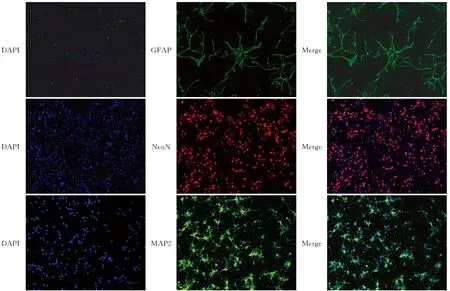
图3 原代神经元和星形胶质细胞的鉴定
3 讨论
在神经系统疾病研究中,神经元和星形胶质细胞是最重要的两种细胞模型。常规的实验方法是选用不同时段的乳鼠分别进行神经元与星形胶质细胞的提取培养。提取神经元选用胎鼠或24 h新生乳鼠[1],个别研究也选用成年鼠;而提取星形胶质细胞选用出生2~3天乳鼠[2]。对于同时研究神经元和神经胶质细胞的实验来说,选用不同时段的乳鼠费时费力,且造成实验动物的浪费。本实验用24 h新生SD大鼠,实现了神经元和星形胶质细胞的同时提取,且获得了满意的结果。另外,将海马和皮层分别处理,一定程度上实现了神经元和星形胶质细胞的分离,减少了后续相应杂细胞的干扰,对于致力于神经系统相关疾病研究的学者来说,提供了一种简便有效的方法。但实验过程中需要注意诸多细节均会对培养效果产生影响。
首先是培养组织的选择。神经元主要存在于大脑皮层和海马,多数研究者用大脑皮层来提取神经元进行培养,但大脑皮层组织除神经元外,还有大量的神经胶质细胞、结缔组织等杂细胞。如果单独用皮层提取神经元,需要在培养后期加入阿糖胞苷抑制非神经元的生长。张余等[3]的研究结果显示阿糖胞苷虽然对神经元产量及形态影响不大,但会明显缩短神经元的存活时间,影响长期培养神经元的存活率。另外有学者提出只取大脑皮质表层2.0~3.0 mm,培养的细胞纯度达90%以上,明显高于取整个皮质的,且该方法不需使用阿糖胞苷[4],但该培养方法所得细胞纯度有待进一步验证。而海马组织含有的神经元种类比较单一,且含量多,有典型的细胞表型,因此常被用来作为提取原代神经元的首选[5]。故本实验中也只选择海马来进行原代神经元的培养,结果证明海马组织培养的神经元存活率较高。另外培养过程中还需注意细胞的纯化。一种纯化神经元的方法是使用无血清培养基Neurobasal+B27(NB),该方法利用血清营养缺失而抑制非神经元的生长[6]。用无血清培养基的方法有两种,一种是10%DMEM/F-12种植培养+NB维持培养;另一种是直接用NB培养。王静欢等[7]的研究结果表明,7天内单纯用NB获得的神经元纯度和活性高。因此,本实验选择单纯用NB来纯化神经元。除此之外,一些操作细节也会影响细胞存活率和活性,如提取细胞时采用的机械吹打和酶消化法。单纯用机械吹打[8]、单纯用酶消化法[2]、机械吹打结合酶消化法[9]均可以提取到满意数量的细胞。需要注意的是,机械吹打时动作一定要轻柔,且分层吹打会减少细胞损伤,提高细胞存活。如果选择酶消化法,木瓜蛋白酶作用温和,获得的结果较为满意[10]。另外有实验室采用0.125%胰酶消化20 min,而本课题组的多次对比研究发现,木瓜蛋白酶消化后神经元存活率高。接种密度也会影响接种效果,接种密度太低,会导致神经元生长缓慢,难以形成神经元网络;接种密度过高,会造成神经元接触抑制、争夺营养,影响神经元的发育成熟[11]。本课题组总结数次实验得出,种植密度在106~108/mL得到的神经元生长最为良好。
由于大脑皮层含有的胶质细胞数量较多,星形胶质细胞的培养相对神经元来说较为容易。其关键在于排除杂细胞的干扰和分离不同的胶质细胞。通过差速离心和梯度血清法可以去除成纤维细胞和内皮细胞,恒温摇床法可将星形胶质细胞与其他胶质细胞分离,神经元的影响可在细胞传代后得到解决。传统的恒温摇床法[12]需要37 ℃恒温摇床,若实验室无此条件,则需将培养瓶口封闭12~24 h在培养箱外摇晃培养,此法会造成细胞的缺氧死亡。丁娟等[2]的方法较为简单,在细胞培养的第7天,通过“十字手摇法”摇晃5 min,也能将星形胶质细胞和其他胶质细胞分离开来。本文在其基础上结合其他方法[13-14]对实验条件进行了优化,得到纯度满意的星形细胞。此外,在实验过程中观察到一些因素会影响细胞纯度和活性。首先是小鼠杀菌消毒时,75%乙醇浸泡时间不宜过长(<5 min),以免导致皮层细胞坏死自融。其次在剥离大脑软脑膜和血管时一定要尽量剥离干净,如可将大脑皮层置于无菌滤纸上轻轻滚动1周,此时脑膜和血管会得到很好的剥离,此步在纯化中意义重大。添加胰酶时,每2只乳鼠加入500 μL胰蛋白酶消化20 min。差速贴壁时间需控制在20 min,若时间过短,成纤维细胞去除效果差;时间过长,有部分星形胶质细胞也会贴附于试管壁,造成提取细胞过少。最后,种植密度也较为重要,实验发现106~108/mL的细胞密度可获得生长良好的细胞。
总之,本课题组通过多次实验验证,使用同一批出生24 h乳鼠分别分离海马与皮质来进行原代神经元与原代星形胶质细胞的同时培养是一种节约、省时、简便、可行的方法,所培养的细胞完全满足后续实验需求。
猜你喜欢
中国实用神经疾病杂志(2022年3期)2022-11-28
世界中医药(2022年17期)2022-10-15
中国应用生理学杂志(2022年2期)2022-08-29
传染病信息(2022年2期)2022-07-15
安徽医科大学学报(2022年6期)2022-07-13
中华骨与关节外科杂志(2022年4期)2022-06-30
山西医科大学学报(2021年5期)2021-06-18
新生代(2019年3期)2019-11-14
初中生世界·七年级(2018年7期)2018-09-07
飞碟探索(2015年11期)2015-09-10
