
狼疮性肾炎足细胞损伤的机制研究进展
2022-01-18 09:54王成玉综述胡伟新审校
肾脏病与透析肾移植杂志 2021年6期
王成玉 综述 胡伟新 审校
狼疮性肾炎(LN)是系统性红斑狼疮(SLE)的严重并发症,肾病范围蛋白尿通常预示着增生性LN(Ⅱ/Ⅲ/Ⅳ级)和(或)膜性狼疮肾炎(Ⅴ级,伴或不伴有Ⅱ、 Ⅲ级或 Ⅳ级病变)的存在。LN 的发病机制是以抗双链 DNA(ds-DNA)抗体等各种自身抗体与抗原结合形成的免疫复合物(IC)沉积以及炎症细胞浸润为主要特点。足细胞是IC的主要攻击对象之一。在各种原因引起的肾小球损伤中,足细胞亦可作为免疫细胞发挥固有免疫及适应性免疫功能,主动参与疾病发展过程。
LN的足细胞损伤
不同类型LN足细胞损伤的特点
Ⅲ型、Ⅳ、V型LN 抗ds-DNA抗体可诱导补体激活和炎症反应, Ⅲ型和Ⅳ型狼疮性肾炎与其有关;膜性狼疮肾炎(Ⅴ型)的特征是上皮下免疫复合物沉积,导致局部补体激活和足细胞功能障碍[1]。
Ⅴ型LN和增生性LN蛋白尿的复杂病理机制可能是不相同的。在增生性LN中,出现足细胞损伤,在Ⅴ型LN中则表现为足细胞发生功能性(非器质性)病变,可能是其长期预后较好的原因[2]。
研究发现增生性LN(Ⅲ型和Ⅳ型)肾小球内皮细胞损伤评分与足突宽度(FPW)密切相关。体外实验表明,LN时IgG刺激足细胞,导致细胞骨架重新分布,血管内皮生长因子A(VEGF-A)水平降低。足细胞暴露刺激细胞外基质后,nephrin减少,可引起细胞骨架重分布。VEGF-内皮素系统可能在LN的发生发展中起着重要作用,它可能是LN新疗法的靶点[3]。
狼疮足细胞病 近来研究发现,少数临床表现为肾病综合征(NS)的 LN 患者,肾活检光镜下仅见肾小球轻微病变(Ⅰ型)或系膜增生性病变(Ⅱ型),免疫荧光及电镜检查IC仅沉积在肾小球系膜区,毛细血管袢内皮下或上皮侧无免疫沉积物,但电镜检查肾小球足细胞足突广泛融合。狼疮足细胞病是一种由于 SLE 与足细胞损伤之间存在密切的病理生理联系造成的特殊类型。
我们报道了50例狼疮足细胞病患者,占LN的1. 33%[4]。目前狼疮足细胞病缺乏统一的定义和诊断标准。肾活检组织除进行光镜和免疫荧光检查外,需依靠电镜检查明确足细胞病变,并排除早期Ⅴ型,甚至Ⅳ型LN,因此,电镜检查是诊断狼疮足细胞病的关键[5]。
研究表明肾小球足细胞是免疫活性细胞,参与T细胞共刺激的B7-1,通过TLR-4激活,被脂多糖(LPS)上调[6]。足细胞摄取可溶性和颗粒性抗原,激活CD4+T细胞,并将主要组织相容性复合体(MHC)Ⅰ类分子上的外源抗原交叉呈递给CD8+T细胞,足细胞也充当抗原提呈细胞,参与免疫介导的肾小球疾病[7]。
蛋白尿与足细胞损伤的关系足细胞损伤与蛋白尿水平相关,研究表明非增生性和增生性LN足突消失融合(FPE)与严重的蛋白尿相关,成熟足细胞的标志物包括突触素、nephrin和肾小球上皮蛋白1 (GLEPP1)等,在增生性LN中丢失[2]。足细胞还可以通过分泌促炎细胞因子[白细胞介素1β(IL-1β),肿瘤坏死因子α(TNF-α),干扰素α(IFN-α)和IFN-γ]来促进LN的炎症反应[8]。T细胞诱导的骨桥蛋白(OPN)可以诱导巨噬细胞向肾小球内浸润[9],OPN可能通过调节足细胞信号和运动,在蛋白尿的发生发展中发挥重要作用[10]。
足细胞损伤的标志物和足细胞抗原
足细胞是LN中IC沉积物的直接或间接靶标。在NZB/WLN狼疮小鼠模型,肾小球足细胞标志nephrin和podocin表达下调[11],表明足细胞功能障碍在LN肾小球病变发生中可能发挥了作用。活动性LN患者尿沉渣的研究发现,podocalyxin,nephrin,podocin和突触足蛋白mRNA的水平与狼疮活性相关[12],活动的LN患者尿中存在未分化nephrin和GLEPP1的足细胞,其蛋白水平与LN蛋白尿和组织学特征相关,尿液足细胞标志物表达可能是评估SLE肾小球疾病进展的潜在有用的无创性标志物。
通过特定的抗原决定簇(特别是膜联蛋白Ⅱ),肾小球系膜细胞和足细胞被确定为IC的直接靶标[13]。此外,已将几种足细胞抗原鉴定为LN中潜在的自身抗体靶标。针对LN患者肾活检样本的蛋白质组学研究发现,肾小球IgG可以识别足细胞抗原,例如α烯醇酶和膜联蛋白A1 (annexin A1)[14]。
足细胞衍生微粒(MPs)是SLE伴LN患者临床和组织学特征的新生物标记物。测定各组尿膜联蛋白(annexin V)、足细胞标记蛋白(podocalyxin)及MPs水平的变化可能有助于评价和监测SLE的活动性[15]。
LN患者nephrin表达显著减少,肾组织巨噬细胞明显增多,肾组织中nephrin的表达与巨噬细胞的浸润呈负相关。因此,LN肾组织中巨噬细胞浸润可能是狼疮肾炎足细胞损伤的重要机制之一[16]。
泛素C末端水解酶L1(UCH-L1)在正常肾脏足细胞中不表达,但在LN的肾小球足细胞中表达明显升高。LN中的核因子κB(NF-κB)/UCH-L1信号传导,包括TNF-α在内的各种刺激均可诱导NF-κB信号通路的激活。NF-κB通过结合其启动子的-300和-109-bp位点来上调UCH-L1表达,导致足细胞的损伤,进一步导致狼疮性肾炎[17]。
足细胞损伤的机制(图1)
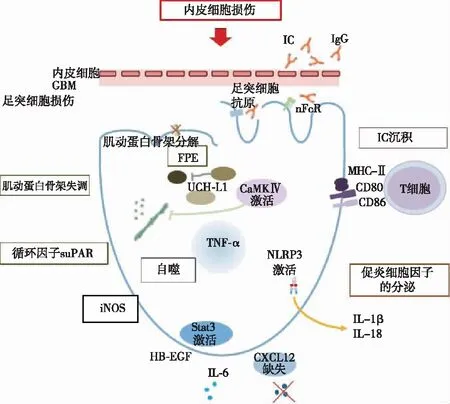
图1 狼疮性肾炎足细胞损伤的机制[1]GBM:肾小球基膜;IC:免疫复合物;nFcR:抗体的Fc受体;TPE:足突融合;TNF-α:肿瘤坏死因子α;CaMKⅣ:钙调蛋白激酶Ⅳ;iNOS:一氧化氮合酶;IL:白细胞介素;HB-EGF:肝素结合性表皮生长因子;suPAR:可溶性尿激酶纤溶酶原激活物受体;NLRP3:核苷酸结合寡聚化结构域受体3;Stat3:信号传导与转录激活因子3;CXCL12:C型趋化因子配体12
免疫复合物沉积LN的组织学分类基于肾小球中的IC沉积物、系膜和(或)毛细血管内病变。然而有证据表明,肾小球上皮细胞,特别是足细胞也与SLE患者的肾小球损伤有关。
IC沉积的部位及其引发炎症是LN肾小球损伤机制的基础。IC通过多种机制引发肾脏损害,沉积在基膜内皮侧的复合物会损伤内皮细胞,是Ⅲ型和Ⅳ型增生性LN的标志。在Ⅴ型LN中发现的上皮侧沉积物损伤足细胞,但由于它们接触原尿而非血液,引发的炎症反应较轻。如果肾小球基膜破裂,这些IC可以进入整个肾小球。免疫沉积物可能启动补体活化级联酶促反应,或直接激活肾小球细胞,诱导炎性趋化因子和细胞因子的释放[18]。在不同的LN类型中均可见足细胞损伤,在增生性LN组中,足突宽度(FPW)与肾脏损伤指标呈正相关,包括蛋白尿水平、血清肌酐以及肾脏病理活动和慢性指标,Ⅴ型LN的FPW较宽,大量蛋白尿的患者表现出更宽的FPW,这表明Ⅴ型LN的蛋白尿水平可能与足细胞功能障碍有关[19]。
内皮细胞损伤研究表明,足细胞和内皮损伤可导致增生性LN,并以NS为表现。足细胞损伤可导致膜型 LN,并表现为NS[20]。与Ⅴ型LN相比,Ⅲ/Ⅳ型LN中基膜与内皮的间距增宽,内皮细胞损伤更明显。肾病范围蛋白尿的足细胞功能障碍也可能以与遗传或自身免疫补体功能失调相关的非典型溶血性尿毒症综合征(aHUS)形式出现。aHUS可能使肾小球磷脂复杂化,这可能是由于足细胞功能异常导致内皮损伤和血栓所致[21]。
可溶性尿激酶纤溶酶原激活物受体(suPAR)suPAR是一个生物标志物,可用于识别在疾病的前5年中有遭受器官损伤风险的SLE患者[22]。suPAR可导致足细胞损伤、足突消失和蛋白尿。suPAR与足细胞膜上的β3整联蛋白的结合会影响足细胞的功能,破坏肾小球的屏障功能,循环中的suPAR升高可诱导足细胞β3整合素活性,从而导致肾脏损伤[23]。suPAR通过与足细胞整合素结合,诱导足细胞迁移和凋亡,参与肾脏疾病的发病进程,被认为是导致 FSGS 的循环致病因子之一[24]。suPAR是LN与其他肾小球疾病足细胞损伤的共同机制之一。
诱导型一氧化氮合酶(iNOS)研究表明,LN的足细胞可表达iNOS,iNOS产生的一氧化氮(NO)可以引起足细胞病,NO可能通过缺氧诱导因子1α和细胞分裂控制蛋白42和Ras相关的C3肉毒杆菌毒素底物1途径诱导这种表型。这项研究表明iNOS可能诱发自分泌足细胞功能障碍[25]。因此,靶向iNOS或其诱导途径可能带来治疗LN的益处。
自噬研究表明足细胞自噬活性是肾损伤的关键因素,几种自噬活性生物标志物可能在疾病早期调节足细胞自噬。LN在培养的足细胞中诱导环氧化酶2(COX-2)和内质网转录激活因子4(ATF4ER)应激途径。抑制COX-2可抑制LN诱导的足细胞自噬。ATF4抑制LN诱导的COX-2过表达。由ATF4ER应激途径诱导的COX-2过表达有助于LN诱导的肾脏自噬和损伤,COX-2可能是LN的潜在治疗靶标[26]。
遗传学机制遗传学机制可能在LN的发展中起主要作用。非裔美国人人群中载脂蛋白L1(ApoL1)的两个常见变异等位基因最近被确定为局灶节段性肾小球硬化症(FSGS)和终末期肾病(ESRD)的危险因素[27]。ApoL1相关足细胞损伤的潜在机制仍不清楚。有人提出,ApoL1变异体参与了改变的囊泡运动,自噬体破坏和自噬障碍,导致足细胞丢失[28]。在LN中观察到的干扰素α过度生产可能构成第二次打击,在存在ApoL1风险等位基因的情况下引发足细胞损伤[29]。
不同的免疫抑制剂对LN足细胞损伤的保护机制
LN的早期足细胞病可出现足细胞裂隙膜蛋白nephrin和podocin的减少,研究者通过免疫印迹和免疫荧光法评估了NZB/WLN狼疮小鼠肾小球nephrin和podocin的表达和定位。治疗三个月后,糖皮质激素和环磷酰胺组的nephrin和Podocin蛋白表达均显着增加。这项研究表明,在LN中用糖皮质激素或环磷酰胺早期治疗可保护足细胞的隔膜蛋白,从而增加了这些药物对足细胞直接保护作用[30]。
免疫抑制剂还可能直接影响足细胞的结构和功能,糖皮质激素可以稳定肌动蛋白,增加肌动蛋白聚合,激活细胞骨架相关的细胞内激酶,抑制IL-6,诱导nephrin表达,从而保护足细胞。研究表明,环孢素A和他克莫司可通过减少足突融合、恢复足突和podocin的机制,减少蛋白尿[31]。
综上所述,多种机制可能参与了LN的足细胞损伤。SLE 时是否存在针对足细胞结构蛋白分子的抗体导致足细胞损伤,某些因子是否通过调节特异性足细胞蛋白表达促进足细胞损伤,是否存在特异性狼疮的足细胞损伤标志物,以及遗传因素是否参与了狼疮足细胞病的发生等问题还有待进一步研究,其有效治疗方案仍有待进一步研究探索。
猜你喜欢
流程工业(2022年1期)2022-11-25
基层中医药(2022年3期)2022-07-22
现代临床医学(2022年2期)2022-04-19
昆明医科大学学报(2021年12期)2021-12-30
老友(2021年3期)2021-03-28
中华养生保健(2020年10期)2021-01-18
家庭百事通·健康一点通(2020年11期)2020-11-30
健康必读(上旬刊)(2019年12期)2019-10-21
安徽医专学报(2018年3期)2018-07-11
现代养生·下半月(2017年12期)2017-10-21
