
Ⅳ型胶原与肾脏疾病
2022-01-18 09:54综述俞晓敏审校
肾脏病与透析肾移植杂志 2021年6期
汪 卿 综述 俞晓敏 审校
Ⅳ型胶原是一种基膜中的细胞外基质蛋白,相互连接的Ⅳ型胶原是肾小球基膜(GBM)中的主要结构成分,对维持肾小球滤过屏障的功能有着重要的作用[1]。Ⅳ型胶原相关的基因突变会破坏GBM形态结构的完整性,进而导致肾脏疾病的发生和进展。随着基因检测技术的进步,特别是新一代测序技术(NGS)的广泛应用, 越来越多的研究拓展了Ⅳ型胶原基因的突变以及与其相关的肾脏疾病谱之间的关系。本文就Ⅳ型胶原基因突变导致相关肾脏疾病的研究进行综述,揭示Ⅳ型胶原相关基因突变的临床表型图谱的演变及对临床的意义。
Ⅳ型胶原在GBM的结构组成及功能
目前已经证实有6种α链(α1-α6)参与组成了Ⅳ型胶原网状结构,编码α1、α2链的COL4A1、COL4A2基因位于染色体13q34上,编码α3、α4链的COL4A3、COL4A4基因位于染色体2q35-37,而编码α5、α6链的COL4A5、COL4A6基因位于X常染色体Xq22[1]。每条α链由三部分组成:由甘氨酸-X-Y重复序列组成的胶原结构域、N端的7S结构域以及C端的非胶原结构域(NC1)[1]。Ⅳ型胶原的形成从α链NC1相互缠绕形成三聚体开始并终止于7S结构域的结合完成。理论上6种α链相互缠绕可形成多种三聚体,然而NC1的结合具有一定的选择性。根据序列的相似性,6条α链可以分为α1型链(α1,α3,α5)和α2型链(α2,α4,α6)[2]。目前发现的α链三聚体存在的三种组合形式(α1α1α2,α3α4α5,α5α5α6)均为两种α1型链与一种α2型链组成。而由一种α1型链与两种α2型链组成的三聚体,因为空间位阻效应,蛋白结构不能稳定存在。
Ⅳ型胶原三聚体的互相连接对维持网状结构单元的稳定性有着重要的作用(图1)[3]。网状结构单元始于NC1的结合,α1α1α2链和α3α4α5链均可与同源三聚体连接,而α5α5α6链不能和自身相结合,只能与α1α1α2链相结合。因此目前存在三种NC1的结合方式,分别为α1α1α2-α1α1α2,α3α4α5-α3α4α5以及α1α1α2-α5α5α6。氯离子在NC1的结合过程中有重要的作用,有实验证实氯离子缺失会影响NC1结合和Ⅳ型胶原网状结构形成[4]。而NC1主要通过其域内的甲硫氨酸残基和羟赖氨酸残基结合形成硫亚胺键相互连接[5],硫亚胺键生成过程中需要离子溴化物和过氧化物酶的催化。相互结合的NC1之间可以形成1个或2个硫亚胺键,因此一个NC1六聚体最多可以存在6个硫亚胺键。动物研究证实过氧化物酶缺失或缺乏溴化物饮食造成硫亚胺键形成障碍会导致基膜结构异常和相应器官发育障碍[6]。在过氧化物酶敲除硫亚胺键减少的小鼠模型中也发现肾小管基膜的结构硬度下降[7],提示Ⅳ型胶原内硫亚胺键对维持基膜机械性功能有着重要的作用。每四条α链7S端通过赖氨酰氧化酶样蛋白2(LOXL2)的调节相互结合形成十二聚体。7S端富含较多的半胱氨酸及赖氨酸残基,其十二聚体的稳定性主要靠α链三聚体内部和彼此之间的二硫键以及羟基赖氨酸交联来维持。α3链和α4链的7S端中有更多的半胱氨酸[8],因此富含更多二硫键的α3α4α5链网状结构比α1α1α2链更加稳固,难以被蛋白酶水解。

图1 Ⅳ型胶原在基膜上的结构与组成[3]
Ⅳ型胶原各亚型间的生化特性和特定细胞表面受体传导的细胞内信号通路导致了不同的生物学功能。Ⅳ型胶原的NC1端和胶原域通过整合素和非整合素受体与细胞表面结合,α1的NC1与整合素α1β1结合,α2的NC1与整合素αvβ3、αvβ5、α3β1结合,α3和α6的NC1与整合素αvβ3结合[8]。α1α1α2胶原域包含整合素α1β1和α2β1的结合位点,借此调节与内皮细胞的粘附和迁移。Ⅳ型胶原同样可以与盘状结构域受体1(DDR1)结合。通过与特异性受体的结合,Ⅳ型胶原各种α链行使了不完全相同的功能。例如α1链和α5链均属于α1型链,但在肺癌患者发现只有α5链的缺失会造成肺癌细胞及内皮细胞中DDR1表达的下降[9],α5链与DDR1的相互作用是激活DDR1及其下游信号通路的关键,因此可以认为DDR1是α5链的特异性受体。Ⅳ型胶原每条α链中胶原域的甘氨酸-X-Y重复序列有一定数目的中断[8]。这些中断点不仅保证了Ⅳ型胶原的弹性,还可以作为链与链、或链与细胞受体之间的结合点。和α1链(21个)相比,α4链(26个)和α6(25个)链具有较多的胶原区中断点,因此α3α4α5链和α5α5α6链拥有比α1α1α2链更强的结合性能。
Ⅳ型胶原网状结构并不是静态结构,存在动态重塑的生理过程。在GBM中发现α1α1α2链存在于内皮下而α3α4α5链存在于上皮侧,α1α1α2链降解并被α3α4α5链替换为是肾脏发育过程中一个至关重要的转变。在缺乏α3链或α5链的动物模型中发现α1α1α2链在肾脏中未被降解,持续存在。在正常的肾脏中,有少量的α1α1α2链局限存在于GBM内皮下,在缺乏Ⅳ型胶原α3链的肾组织中α1α1α2链广泛分布于GBM两侧[10]。虽然α1α1α2链能够替代α3α4α5链的不足组成Ⅳ型胶原的网状结构单元,但由于不同亚型间的结构与功能的差异,α1α1α2链不能够完全代偿因α3α4α5链缺乏导致的IV胶原在肾脏中的功能缺失,进而引发了一系列肾脏病。
Ⅳ型胶原基因突变相关疾病
Alport综合征Alport综合征是一种临床表现为血尿、蛋白尿和肾功能不全并常常伴有高频听力的丧失以及眼部病变的进展性遗传性疾病[11]。Ⅳ型胶原COL4A3/4/5基因突变造成α3α4α5链的结构功能异常是Alport综合征的主要病因。电镜下GBM的厚薄不一、断裂分层是此病的典型特征性病理改变。既往根据遗传方式的不同,Alport综合征主要分为三类:COL4A5基因突变引起的X染色体连锁遗传(XLAS),COL4A3/4基因突变引起的常染色体隐性遗传(ARAS)和常染色体显性遗传(ADAS)[12]。XLAS男性患者临床症状较重,COL4A5基因型与XLAS男性患者进展至终末期肾病(ESRD)的年龄相关联,无义突变和造成蛋白截短的剪接突变比携带错义突变的患者病情更重,进展至ESRD的时间更早[13]。对XLAS女性患者大样本临床观察研究表明,XLAS女性患者临床表型复杂多变,并且未发现明显的基因型-表型关联,可能多种不同的机制共同决定了XLAS女性患者疾病严重程度[14]。与XLAS男性患者相似,ARAS患者表型严重,进入ESRD的年纪较小(20岁左右),基因型-表型关联分析发现携带非错义突变的患者比携带错义突变的患者预后更差[15]。携带COL4A3/4基因杂合突变的ADAS患者较为少见,表型较轻,进展至ESRD的速度慢,病变可以局限于肾脏,缺乏其他临床表现。
既往研究认为近85%的Alport综合征为XLAS,ARAS占Alport综合征全部的15%左右,ADAS仅仅占1%左右。随着近年来基因测序技术在临床诊断中的广泛应用,研究表明,实际由COL4A3/4基因突变导致的Alport综合征比例更多,特别是ADAS患者的比例达到了接近30%[16-17]。因此仍需要较大样本的研究来证实三类Alport综合征的比例。另外Mencarelli等[18]报道了COL4A3/4/5双基因致病的病例,提示Alport综合征中存在双基因致病的遗传模式。家系共分离分析提示双基因遗传的Alport主要分为三种:(1)常染色体基因(COL4A3/4)上分别来自于父母的两个突变,临床表型与ARAS相似;(2)常染色体基因(COL4A3/4)来自于父亲或母亲的两个突变,临床表型与ADAS相似;(3)分别来自于父母的常染色体基因(COL4A3/4)和X染色体基因(COL4A5)上的突变,临床表型不明。目前双基因致病案例报道较少,无法归纳此类患者的特点,有待进一步的观察和研究。因此,根据近年来的研究进展,Alport综合征被重新分为X连锁型、常染色体型以及双基因型三类(表1)[19]。
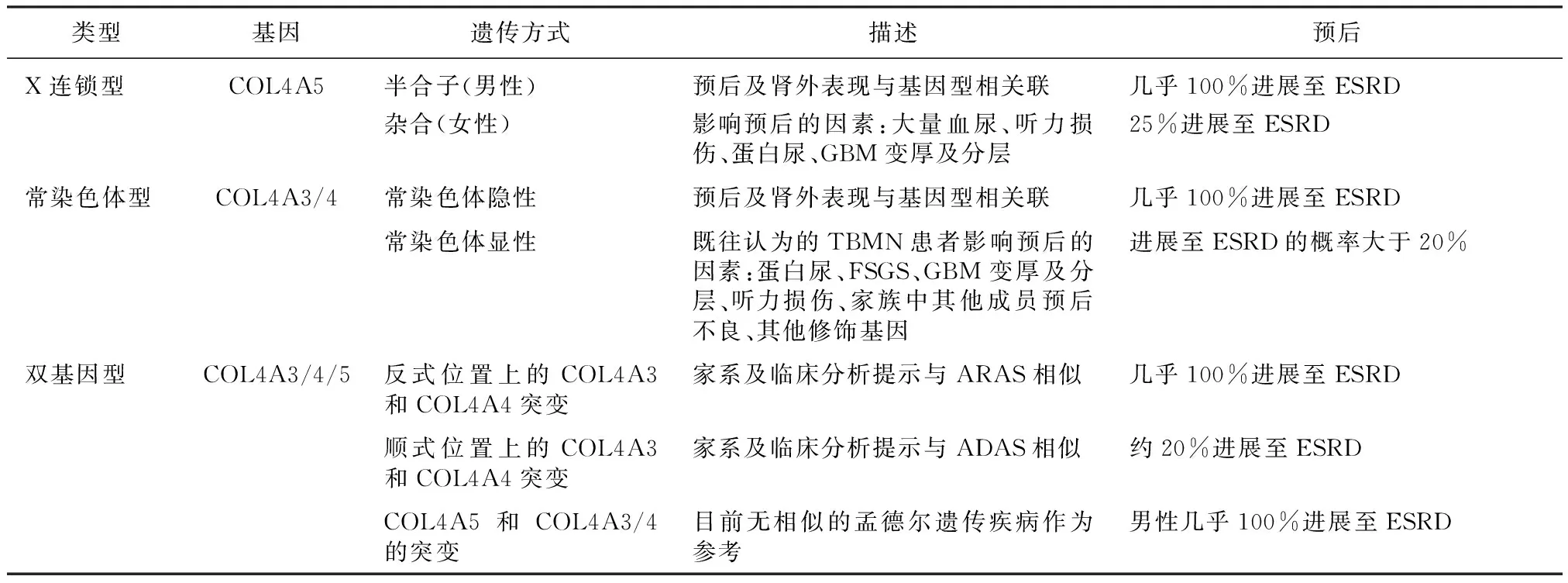
表1 Alport综合征的分型[19]
Alport综合征尚无特效治疗手段,首要治疗目标仍然为延缓疾病的进展。Alport综合征的临床和动物实验证实了血管紧张素转化酶抑制剂(ACEI)和血管紧张素Ⅱ受体拮抗剂(ARB)可以降低蛋白尿的水平,延长患者进展至ESRD的时间,延缓肾脏间质纤维化的发展[20]。在错义突变的Alport综合征患者中,ACEI/ARB延缓患者进展至ESRD的时间长达20年以上[13]。甚至在尚未出现明显蛋白尿的Alport儿童中,ACEI/ARB也可延缓疾病进展,并未出现明显的药物不良反应[21]。以上研究提示,通过基因测序等无创手段尽早明确Alport患者的诊断,及时运用药物干预,对临床上Alport综合征的管理有重要的指导作用。短期FK506治疗也能够能改善Alport综合征患者的蛋白尿及低白蛋白血症,儿童患者疗效可能优于成年患者,且成年患者发生不良反应的概率也可能较高[22]。Wang等[23]已经成功构建携带患者相同突变的Alport综合征类器官模型,为进一步的疾病机制和药物筛选研究提供了有力的工具。目前也已经开展了基因治疗的相关研究,应用外显子跳跃技术可以在携带截短突变的严重表型XLAS小鼠中恢复肾脏基膜区域α5链的表达,重新组成了Ⅳ型胶原三聚体,明显缓解了病变小鼠的临床病理症状[24]。
薄基膜肾病(TBMN)携带COL4A3/4/5基因突变的一部分患者肾组织活检电镜下仅可见到GBM变薄(成人GBM厚度<250 nm,2~17岁儿童<180 nm),无明显断裂分层,故而根据其病理改变被称为TBMN。COL4A3/4基因杂合突变是导致TBMN的一个重要原因[25]。典型的TBMN大多数无明显进展性病变,仅仅表现为孤立性血尿。然而Alport综合征的患者在疾病早期时也可以表现为TBMN样病变。此外,在诊断为TBMN携带COL4A3/4突变的患者中,长期随访的结果表明会出现蛋白尿以及其他类似Alport综合征的临床表现。一项针对COL4A3/4基因突变的META分析结果显示,携带COL4A3/4基因杂合突变的患者中,15.1%的患者在平均52.8岁时进展至ESRD[26],由此可见不能简单认为TBMN是一种良性病变。但与Alport综合征相比,携带COL4A3/4基因杂合突变的肾外临床症状表现较少,如明显的听力下降等。携带COL4A3/4杂合突变的父母,子女出生携带纯合突变的概率为25%,可以表现为典型的Alport综合征。携带COL4A5突变的母亲也同样有50%的几率孕育表型更为严重的男性XLAS患者。即使在同一个家系中,TBMN和Alport均可能同时存在,因此TBMN与Alport综合征不能完全割裂成两个不同的疾病。相较于传统的临床病理诊断,分子诊断可以帮助明确TBMN患者的病因,排查家系中是否存在尚未出现临床表型的突变携带者,指导早期防治。
局灶节段性肾小球硬化(FSGS)FSGS是以部分肾小球节段硬化为特征的病理形态学诊断,电镜下可以看到广泛的足突融合。FSGS患者的临床表现并不完全相同,一部分患者症状较重治疗效果较差,传统的激素治疗对其无效,也被称为激素抵抗型肾病综合征(SRNS)。FSGS是不同病因导致的同一种病理改变,并不是单一疾病。各种原因导致的足细胞损伤被认为是造成典型FSGS样改变的原因[27]。目前已经发现与足细胞功能密切相关的多种基因突变可以导致FSGS[26]。多项关于FSGS的遗传学研究也证实Ⅳ型胶原相关COL4A3/4/5基因突变是成人发病的家族性FSGS常见的致病基因。Gast等[28]在英国76个原发性FSGS的家系中,发现22%有家族史的患者和10%无家族史的患者携带致病突变,COL4A3/4/5基因突变占到了所有突变的56%。Gribouval等[29]在一项包含135例成年散发FSGS/SRNS法国患者的队列中发现43.7%的突变基因为COL4A3/4/5。Yao等[30]在加拿大的一项多种族成人发病FSGS研究发现,11%的患者携带明确的致病突变,其中55%是COL4A3/4/5基因突变,另有9%的患者携带了可疑致病突变,其中37%是COL4A3/4/5基因突变。以上研究中表明,相比于儿童发病的FSGS/SRNS,成人迟发性的FSGS/SRNS中发现COL4A3/4/5基因突变比例相对较高。COL4A3/4/5基因突变可以单独导致FSGS样病变,也可以和足细胞相关基因一起参与FSGS的发生及影响疾病的严重程度。
Ⅳ型胶原相关基因突变导致FSGS的机制尚不完全明确,有假说认为COL4A3/4/5基因突变对肾脏GBM的改变导致了足细胞脱落进而表现为FSGS,也有观点认为COL4A3/4/5基因突变导致异常的α3α4α5链不能有效的组装成Ⅳ型胶原网状结构,游离的Ⅳ型胶原基因相关蛋白在细胞内聚集会造成足细胞的损伤和凋亡[31]。而且在Alport综合征或TBMN的疾病进展过程中,FSGS也可以继发于GBM的损伤[32]。COL4A3/4/5基因的突变在FSGS/SRNS中的发病机制仍不完全清楚,深入研究有助于为FSGS的诊治提供新思路。
小管间质病变除了GBM,Ⅳ型胶原也是组成肾小管基膜(TBM)的主要成分,Alport综合征患者肾组织Ⅳ型胶原染色也发现肾小管部位的表达缺失。虽然有部分的α3α4α5链,但TBM的主要Ⅳ型胶原成分为α1α1α2链[33]。目前,COL4A1/2基因突变主要认为可导致小血管的病变。小鼠COL4A1 基因的突变抑制了突变型和正常型Ⅳ型胶原蛋白的分泌,导致血管基膜的异常组装,从而引起小血管畸形,在部分小鼠内表现为脑内小血管脆性增加、视网膜动脉迂曲、蛋白尿等[34]。COL4A1基因突变引起的肾脏病变报道较少,仅在一种常染色体显性遗传的遗传性血管病、肾病、动脉瘤和肌肉痉挛(HANAC)综合征中被发现,主要表现为脑白质病变、动脉瘤、视网膜动脉病变、血尿、多囊肾、肌肉痉挛等。在HANAC综合征患者发现其GBM和TBM区域内有类似Alport综合征患者的经典GBM断裂分层样改变[35]。然而在一项COL4A1基因突变的动物模型研究中仅在GBM和包囊观察到基膜的结构缺陷,TBM结构无明显变化,COL4A1基因突变的小鼠肾小管功能异常主要是因为慢性内质网应激造成的肾髓质萎缩[36]。因此,关于Ⅳ型胶原相关突变如何导致了TBM的结构功能异常尚未完全清晰,病变是否局限于肾小管内还是全身系统性疾病的一部分,基因突变和表观因素是否共同参与了疾病的发生发展,均需进一步的研究和分析。
综上所述,随着基因组医学的发展,尤其是基因测序技术在临床上的广泛应用,我们有可能从基因层面重新认识疾病,诸如Alport综合征、TBMN及FSGS等以临床表现和病理特征为诊断依据的疾病,究其病因都是由于Ⅳ型胶原相关基因突变导致。目前有作者建议将其统一命名为Ⅳ型胶原肾病。因此,系统开展中国人群Ⅳ型胶原基因相关突变与肾脏疾病之间的大样本队列研究,对于进一步认识由Ⅳ型胶原基因突变导致的肾脏疾病,提高诊断水平,指导用药和随访管理有着重要的临床研究价值和科学研究意义。
猜你喜欢
皮革科学与工程(2022年1期)2022-01-15
河北果树(2021年4期)2021-12-02
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年2期)2021-01-18
世界最新医学信息文摘(2021年6期)2021-01-06
医药前沿(2020年20期)2020-11-10
河北农业科学(2019年6期)2019-03-21
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
