
浊点萃取/超高效液相色谱-串联质谱法测定还亮草中五种生物碱
2022-01-13 12:17樊轻亚王万好
分析科学学报 2021年6期
樊轻亚, 王万好
(1.信阳职业技术学院药学院,河南信阳 464000;2.河南同源制药有限公司,河南信阳 464000)
还亮草(DelphiniumanthriscifoliumHance)属毛茛科、翠雀属多年生草本植物,分布于我国广东、广西、贵州、浙江、江西、河南等省,用于治疗风湿痛、半身不遂、痈疮癣癞等疾病。药用还亮草中的主要有效成分是生物碱类物质,发挥重要的药理作用[1]。因此,研究还亮草中生物碱成分具有较高的临床应用价值。
浊点萃取(Cloud Point Extraction,CPE)是一种新型分离技术,该技术主要利用表面活性剂溶液的增溶和分相实现溶质的分离和富集。浊点萃取不使用挥发性有机溶剂,无毒,操作方便,同时具有很高的富集效率,在中药材有效成分的萃取分离中具有良好的应用前景[2 - 12]。
超高效液相色谱-串联质谱(UPLC-MS/MS)联用技术具有精密度高、准确度好、稳定性好等优点,在中药的定性定量分析中具有广泛的应用[13 - 17]。本文采用浊点萃取方法分离富集还亮草中的生物碱成分,并与UPLC-MS/MS联用测定该药材中5种生物碱的含量,为还亮草的开发和应用提供可靠的依据。
1 实验部分
1.1 仪器、试剂与材料
美国 Waters Xevo TQ-MS型超高效液相色谱-串联质谱仪,配有电喷雾离子源。DFY-200摇摆式高速中药粉碎机(温岭市大德中药机械有限公司)。
洋翠雀碱、高硬飞燕草碱、硬飞燕次碱、硬飞燕草碱、巴比翠雀碱标准品(纯度>98%,成都普菲德生物技术有限公司)。精密称取洋翠雀碱、高硬飞燕草碱、硬飞燕次碱、硬飞燕草碱、巴比翠雀碱5种对照品各10.0 mg,分别用甲醇溶解并定容至10 mL,得各对照品储备溶液。分别吸取储备溶液各1 mL于50 mL容量瓶中,用甲醇定容后即得混合对照品溶液。甲醇、乙腈(色谱纯,德国默克)。实验用水为超纯水。
还亮草药材均购于网络药材市场,经鉴定为还亮草(DelphiniumanthriscifoliumHance)全草。
1.2 实验方法
1.2.1 微波辅助提取称取一定量100目的还亮草药材粉末于提取罐中,按照1∶60的料液比加入95%乙醇后,置于密闭微波瓶中,加热提取(80 ℃,10 min),提取液在60 ℃下挥干,用甲醇定容至200 mL,备用[18]。
1.2.2 浊点萃取取一定量的Triton X-114,加入一定量的水,超声溶解,配制成4%的Triton X-114溶液,加入0.4 g的NaCl振摇,静置,使形成浊点体系。取“1.2.1”浓缩液于离心管中,挥干溶剂,加入配制好的Triton X-114浊点体系溶液,超声溶解,用稀HCl调节pH值为8,55 ℃水浴平衡25 min,分层后在3 000 r/min下离心5 min。取下相置于圆底烧瓶中,挥干,用甲醇定容至10 mL,0.22 μm微孔滤膜过滤后,用UPLC-MS/MS法测定。
1.2.3 溶剂法萃取取“1.2.1”浓缩液减压抽滤,滤液旋干,用30 mL HCl(pH=2)超声溶解,倾出,置于分液漏斗,用氯仿萃取后的水相用1%NaOH溶液调节pH为10,再用氯仿萃取3次,每次30 mL,收集氯仿层,旋干,用甲醇溶解,定容,0.22 μm微孔滤膜过滤,待测。
1.3 样品分析条件
1.3.1 色谱条件色谱柱:ACQUITY UPLC BHE C18柱(50 mm×2.1 mm,1.7 μm;美国Waters公司);流动相:乙腈(A)-0.1%三乙胺(B);梯度洗脱程序:0~3 min,2%~10%A;3~8 min,10%~50%A;8~10 min,50%~70%A; 10~15 min,70%~2%A;柱温:30 ℃;流速:0.3 mL/min;进样量:3 μL。
1.3.2 质谱条件离子源:电喷雾离子源;扫描方式:正离子(ESI+)模式;离子源温度:130 ℃;干燥气温度:350 ℃;干燥气(N2)流速:9.0 L/min;毛细管电压:3.0 kV;锥孔气流量:50 L/h;碰撞气流量:2.5 L/h。
2 结果与分析
2.1 浊点体系的筛选及萃取条件的优化
2.1.1 浊点体系的选择本实验分别选用Triton X-100、Triton X-114、Span-80、Tween-80、 PONPE 7.5、Genapol X-080、油酸聚乙二醇甘油酯和聚甘油脂肪酸酯作为表面活性剂,加入NaCl和一定量的水超声溶解形成浊点萃取体系溶液,水浴加热,测定其浊点温度。结果显示:Tween-80、Span-80、PONPE 7.5分相较慢;Genapol X-080、油酸聚乙二醇甘油酯、聚甘油脂肪酸酯表面活性剂聚集成糊状;Triton X-100、Triton X-114分相较快,表面活性剂富集在下相。但Triton X-100浊点温度为65 ℃,而Triton X-114浊点温度为35 ℃,浊点温度较低,因此选择萃取体系中Triton X-114作为表面活性剂,NaCl作为添加剂进行萃取。
2.1.2 Triton X-114浓度和 NaCl添加量对萃取率的影响本实验考察了不同浓度的表面活性剂和不同NaCl的添加量对还亮草中生物碱萃取率的影响。从图1(a)中可以看出生物碱的提取率随Triton X-114浓度的增大呈现先升后降的趋势。这是因为表面活性剂浓度过低时,萃取分层时表面活性剂富集相过少,无法进行相分离,随着浓度的增大,形成的胶束越来越多,增溶作用越强,越有利于生物碱的提取。但在Triton X-114浓度高于4%之后,提取效率呈下降趋势。因为随着表面活性剂浓度的增大,溶液黏度增大,扩散能力减弱,胶束难渗入到基质的内部,导致胶束内部结构发生变化导致提取率降低。因此,Triton X-114的最佳浓度为4%。图1(b)表明,随着NaCl用量的增加,萃取效率一直呈上升趋势, NaCl用量增加到0.4 g,达到了平衡。这是因为溶液中加入电解质 NaCl 可增大表面活性剂胶团的聚集数,增大溶液离子强度,使水相密度增大,加速两相分离,降低了浊点温度,从而提高对还亮草生物碱的增溶能力。因此选择NaCl的加入量为0.4 g。
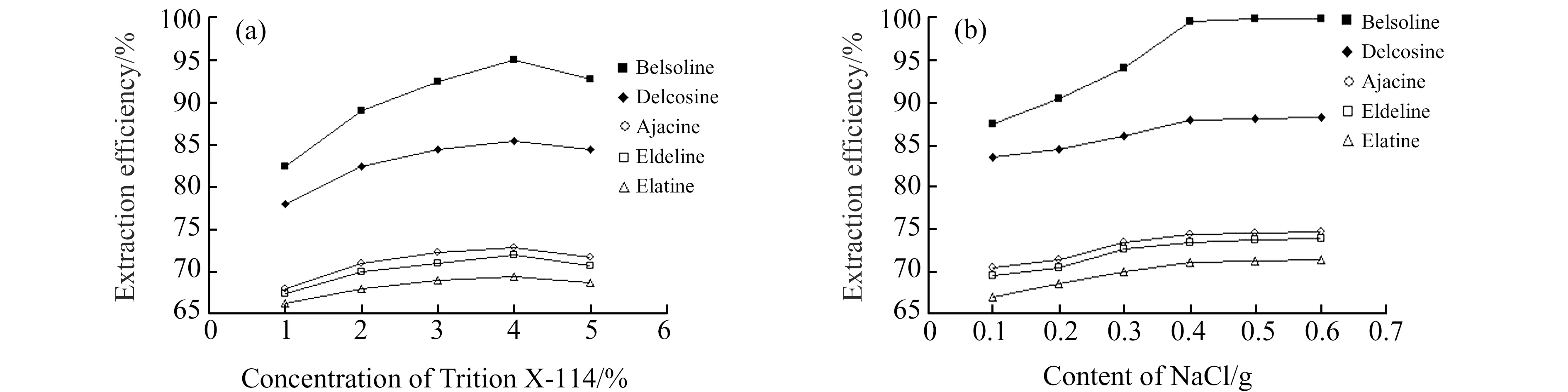
图1 Triton X-114的浓度和 NaCl的用量对还亮草生物碱萃取率的影响
2.1.3 pH对萃取率的影响在浊点萃取中,待测物与非离子表面活性剂胶束以疏水分配为主要方式,样品溶液pH值影响两相分配的效果,通常情况下会将其酸度值调至物质呈电中性状态,易于与胶束结合。本实验中的5种生物碱均显弱碱性,加稀HCl或稀氨水调节pH值至5~10。由图2可知pH为8时,萃取率最大。这是因为在酸性环境下,弱碱性物质易电离成离子状态,不易与表面活性剂结合,难以被胶束萃取,故萃取率较低。而当萃取物pH过高时,使表面活性剂富集相由下相转移至溶液上层,不便于两相分离。因此选择HCl调节pH值为8比较合适。

图2 pH值对还亮草中生物碱萃取率的影响
2.1.4 平衡温度和平衡时间对萃取率的影响本文分别考察了在25~85 ℃水浴中平衡5~30 min对萃取率的影响。结果如图3所示,萃取体系在水浴温度55 ℃下平衡25 min时萃取率达到最大。这是因为当平衡温度低于表面活性剂浊点时,两相系统无法形成;平衡温度上升,在水溶液中,以水合形式存在的表面活性剂会使胶束中氢键断裂脱水,使表面活性剂富集相体积减小,表面活性剂更易沉降,引发更好的相分离,所以平衡温度要稍高于浊点温度约15~25 ℃,当温度过高时,容易引起药物氧化分解,故萃取率下降[19]。延长平衡时间有利于目标物在浊点体系中的分配,当平衡时间为25 min时,萃取率基本达到平稳的状态,若过长的平衡时间反而会增加样品的处理时间,同时会影响实验周期。

图3 平衡温度和平衡时间对还亮草生物碱萃取率的影响
通过单因素试验优化得到了浊点萃取还亮草中5种生物碱的最优条件:4%的Trion X-114,NaCl的添加量为0.4 g,溶液的pH值调节为8,平衡温度55 ℃,平衡时间25 min。
2.2 色谱-质谱条件的优化
2.2.1 色谱条件的优化对还亮草浊点萃取后的提取液进行UPLC-MS/MS分析,考察了有机相溶剂(乙腈、甲醇)与水,以及不同种类的缓冲溶液作为流动相时的分离效果和对离子化程度的影响。结果发现,采用乙腈作为流动相时,分离度更好,且能明显缩短分析时间,而在流动相中加入一定比例的三乙胺可以改善峰形,减少色谱峰拖尾。因此,本实验以乙腈-0.1%三乙胺溶液为流动相,选择梯度洗脱方式,5种生物碱成分可以得到良好分离,且各色谱峰的峰形好、柱效高。所得总离子流色谱图如图4所示。
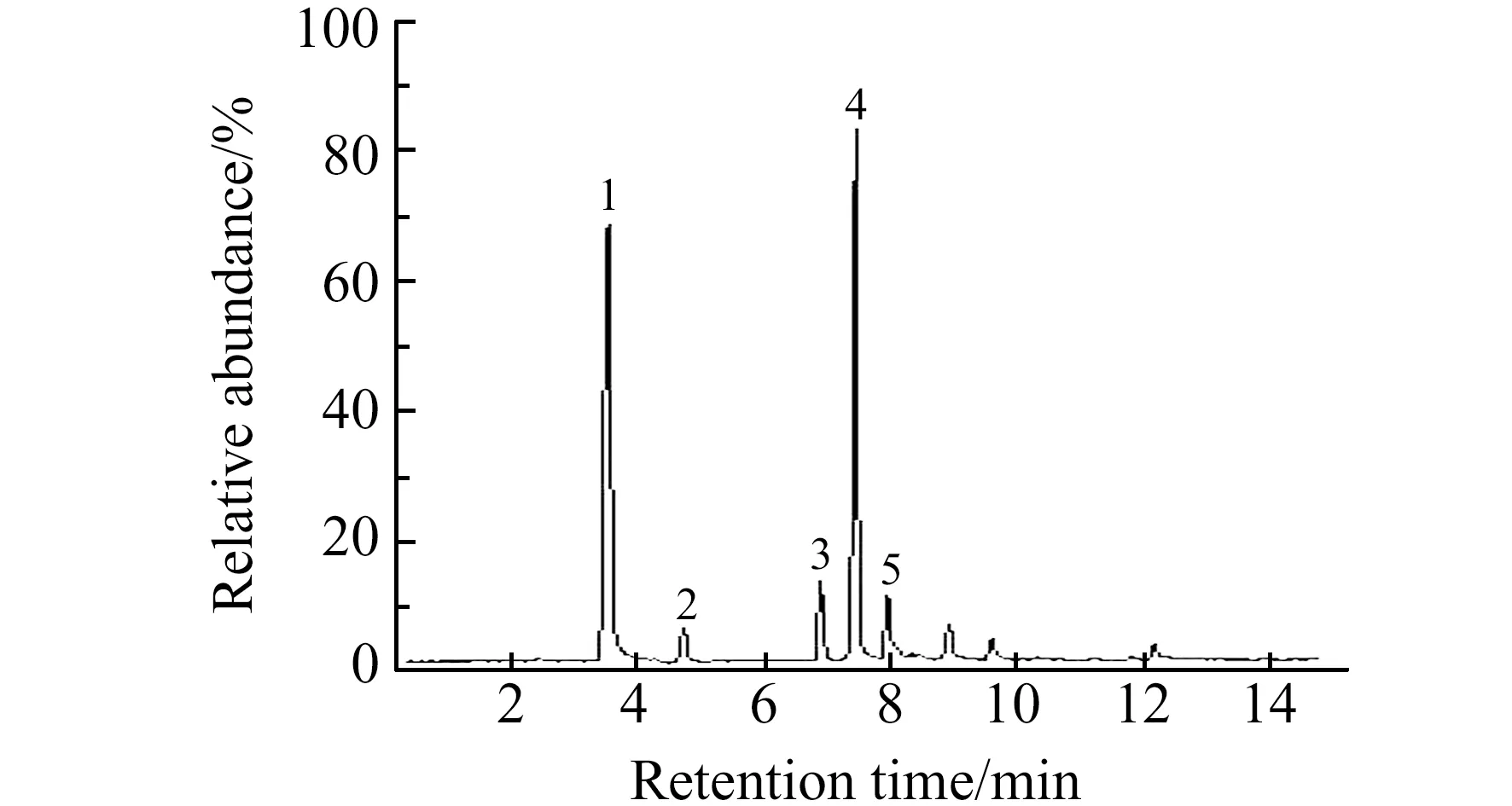
图4 药用还亮草浊点萃取后提取液的总离子流(TIC)色谱图
2.2.2 质谱条件的优化和裂解一级质谱在全扫描模式下,同时采集正、负离子信号,结果发现ESI-模式下几乎无质谱响应,因此选择 ESI+模式,分别找出5种化合物的[M+H]+准分子离子峰。并对干燥气体、雾化器压力、干燥气流量等参数进行优化,然后对母离子进行碰撞诱导解离,选取所得丰度较高的两个子离子作为特征离子,选择响应最高的子离子作为定量离子。5种化合物的质谱参数见表1。

表1 5种生物碱的质谱参数
在ESI+-MS/MS条件下,5种化合物主要发生侧链断裂,根据二质谱级数据,可以推测5种化合物均发生A和B两种裂解路径。5种化合物的 A裂解方式都相同,即16位脱甲氧基;B裂解途径中硬飞燕次碱和硬飞燕草碱发生1位羟基的断裂,高硬飞燕草碱和洋翠雀碱发生4位酯基的断裂,巴比翠雀碱发生6位的脱羧反应。5种化合物质谱裂解途径见图5。

图5 5种生物碱的质谱裂解途径
2.3 线性范围、检出限与加标回收率
取5种生物碱标准溶液,分别配制成一系列浓度的混合标准溶液,按优化浊点萃取及UPLC-MS/MS法分析测定。以目标定量离子的峰面积(Y)为纵坐标,以质量浓度(X,mg/L)为横坐标绘制标准曲线。取已知含量的还亮草样品 3 份,分别添加 0.1 mg/g、0.25 mg/g、0.50 mg/g 3个浓度水平的混合对照品溶液,采用“1.2.1”及“1.2.2”方法进行提取和萃取,在优化色谱和质谱条件下进样定量分析,每个水平做6个平行,计算平均回收率和相对标准偏差(RSD)。5种生物碱的线性方程、线性范围、相关系数、检出限(S/N=3)及加标回收率结果见表2。

表2 5种生物碱的标准工作曲线、线性范围、检出限及加标回收率
2.4 稳定性与重复性实验
取供试样品和混合对照品溶液,于室温下放置0~24 h后,分别于0、4、8、10、12、24 h进样进行测定。结果表明供试样品和对照品溶液在24 h稳定性良好,稳定性和重复性的RSD小于2.5%。
2.5 萃取方法的比较及应用
精密称取3个产地还亮草药材各100 g,按照“1.2.1”方法提取后,分别按照“1.2.3”浊点萃取法(CPE)和“1.2.4”溶剂法(SE)萃取,然后按“1.3”方法测定其含量,结果见表3。由表3可知,3个产地的还亮草生物碱含量分布不同,3个产地中硬飞燕草碱含量最高,广西产地的5种生物碱含量明显高于其它两地,可能和该植物生长时所需的温度与地质条件有关。3批还亮草中采用浊点萃取法的萃取率是传统溶剂萃取法的2倍左右,可能由于传统溶剂萃取法操作复杂,提取后需经过多次萃取,在进行氯仿萃取时每次都需要蒸发溶剂,导致待测物在蒸发及多次的转移过程中损失较多;而浊点萃取法集分离富集与一体,操作简便,和传统溶剂法相比不需要蒸发溶剂,避免了待测物的损失,所以萃取率比传统溶剂法高。且浊点萃取法中所用表面活性剂无毒无害,用量小,降低了污染,且能够保护被萃取物质的原有特性,能够提供较高的萃取率,是一种环境友好型的萃取富集方法,符合绿色化学发展的要求。

表3 3个批次还亮草药材5种生物碱的提取分析结果(n=3)
3 结论
本文将浊点萃取技术应用于还亮草中生物碱的萃取,并优化了超高效液相色谱-串联质谱法测定生物碱的条件。该萃取方法简单快速,萃取效率高,是一种高效的、具有极大发展前景的萃取技术;超高效液相-串联质谱法灵敏度高,分离度好,可以缩短分析时间,满足还亮草药材中5种生物碱成分的同时检测要求,可为还亮草制剂的开发及质量控制提供了可靠依据。
猜你喜欢
皮革制作与环保科技(2022年18期)2022-11-26
新材料产业(2022年2期)2022-07-19
现代仪器与医疗(2022年1期)2022-04-19
中草药(2022年5期)2022-03-03
现代仪器与医疗(2021年2期)2021-07-21
昆明医科大学学报(2021年2期)2021-03-29
当代化工(2019年4期)2019-12-03
商情(2019年43期)2019-10-20
分析化学(2019年3期)2019-03-30
分析化学(2018年12期)2018-01-22
