
表面氧化态可调的α-MnO2纳米线选择性催化氧化5-羟甲基糠醛
2022-01-10 03:09赵婉娜周春梅靳玉广杨艳辉
化工学报 2021年12期
赵婉娜,周春梅,靳玉广,杨艳辉
(南京工业大学化学与分子工程学院,江苏 南京 211816)
引 言
生物质衍生化合物5-羟甲基糠醛(HMF)是连接生物质与化工生产的桥梁分子,通过催化氧化反应可以得到高附加值的2,5-呋喃二甲醛(DFF)、2,5-呋喃二甲酸(FDCA)、5-甲酰基-2-呋喃酸(FFCA)等在化工、医药及燃料领域皆有重要应用的平台分子。其中,具有对称分子结构的DFF可用于合成生物聚合树脂、席夫碱、药品、大环配体、荧光剂等重要化学品[1-4]。目前,经济高效生产DFF的方法是以氧气作为氧化剂,在甲苯、DMF、DMSO等有机溶剂[5]中对HMF进行催化氧化,所使用的催化剂主要包括钒基[6-10]、钌基[11-14]和锰基[15-20]催化剂。聂俊芳[21]通过对比研究发现,在HMF选择性氧化制备DFF的各种非均相催化剂中,锰基催化剂普遍比钒基和钌基催化剂展示出更高的活性和稳定性,主要原因是:锰基催化剂的活性物种通常以化合物形式存在,具有更好的稳定性;相比之下,对于钌基贵金属催化剂和钒基过渡金属催化剂,催化剂的浸出和颗粒大小更容易使钌基和钒基催化剂失活。此外,锰基催化剂由于具有价格低廉、无毒无害、环境友好等特点,被认为是一种更有潜力的催化剂,在HMF选择性氧化反应中的应用也受到越来越多的关注。Lin等[15]研究了不同种类的MnO2晶体催化氧化HMF生成DFF,在100℃的DMSO溶剂中反应3h,其中β-MnO2具有很好的催化活性,DFF产率高达97%,并且该催化剂循环利用5次以上没有明显失活。Chen等[16]以MnOx/P25-600-5h作为催化剂,在30bar(1bar=105Pa)的空气压力下,在140℃的乙醇水溶液进行HMF氧化,反应2h达到转化率33.2%和97%的DFF选择性。Ke等[17]发现氮掺杂MnO2后,使得Mn—O键略微伸长,Mn—O配位数下降,从而产生更多的表面缺陷位点和配位不饱和Mn位点,使催化剂催化HMF氧化成DFF的活性显著提高。以甲苯为溶剂,在室温、0.1 MPa O2条件下反应6h,HMF的转化率达到100%和99%的DFF选择性。以含磁性的Fe3O4负载Mn3O4得到的Mn3O4/Fe3O4催化剂也被应用于HMF选择性氧化制备DFF,以DMF为溶剂在120℃温度下反应12h,可使HMF完全转化且DFF的产率可达到82.1%[18]。2019年,Lin等[19]发现Fe2O3可以提高MnO2氧化HMF生成DFF的活性,他们合成的Mn6Fe1Ox混合氧化物催化剂在110℃、1.5 MPa O2压力下,以DMF为溶剂反应6h可以得到97%的HMF转化率和96%的DFF选择性。
此外,大量研究结果表明,多种具有特殊结构的锰基催化剂都能高效地催化HMF制备DFF,一些研究者对它们的催化机理进行了探索。根据Suib等[22]的报道,OMS-2催化C—H键的活化可能经历Mars-van Krevelen过程,Nie等[23]利用同位素实验,证实K-OMS-2催化剂上晶格氧参与到了HMF氧化反应中,在他们提出的反应机理中(图1),反应的第一步是晶格氧与HMF反应生成DFF,同时导致Mn4+被还原成Mn3+;然后,被消耗的晶格氧会被分子氧填充,同时伴随着Mn3+被再氧化成Mn4+,从而完成氧化还原循环。Florea等[20]也报道了锰铜层状双氢氧化物(Mn0.70Cu0.05 Al0.25 )在水相中催化HMF生成DFF的反应机理,该反应是以Cu2+转化成Cu+为第一步进行的,随后引发了Mn4+和Mn3+的氧化还原循环。
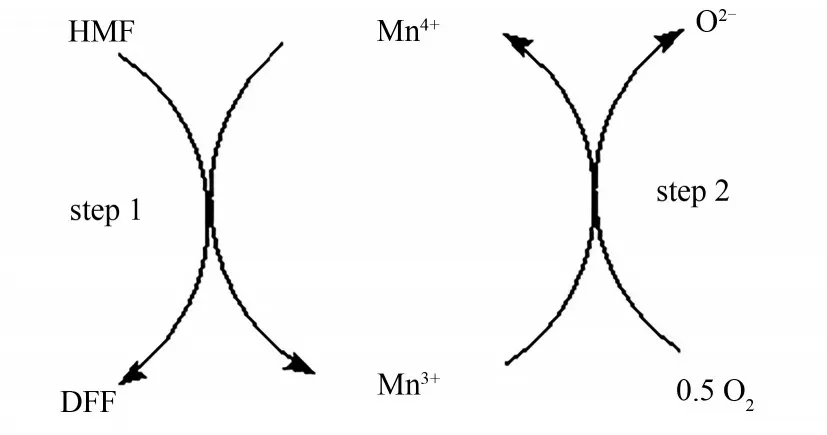
图1 K-OMS-2选择性氧化HMF的机理[23]Fig.1 Catalytic mechanism of aerobic oxidation of HMF over K-OMS-2[23]
上述机理研究表明,氧化态可调变的Mn物种是重要的催化活性中心,在反应过程中动态变化的Mn4+、Mn3+甚至Mn2+物种会对催化过程产生重要的作用。本文利用水热法制备一系列α-MnO2催化剂,通过改变水热合成条件、氧化/还原气氛焙烧处理等方法,对催化剂中Mn4+、Mn3+、Mn2+物种的含量进行调变,并对催化剂的平均氧化态(AOS)和催化活性的关系进行系统的研究。
1 实验方法和表征
1.1 纳米MnO2催化剂的合成
将0.2mmol KMnO4、0.2mmol NH4Cl与40ml超纯水经30min超声混合后置于水热反应釜中,加热至140℃后恒温保持24h,取出冷却后,过滤出固体物质,使用超纯水清洗后在100℃烘干过夜得到粉末催化剂样品,命名为MnO2-NF。对水热温度和时间进行调变,改为170℃恒温24h,200℃恒温24h和40h,得到的样品分别命名为MnO2-NW1、MnO2-NW2、MnO2-NW3。此外,从阿拉丁购买的商业二氧化锰(纯度>90%)也被用作催化剂进行比对,在文中命名为MnO2-NR。
1.2 结构表征
扫描电镜(SEM)表征在FEI INSPECT S50场发射电子显微镜上进行,加速电压为15kV。透射电镜(TEM)和高分辨透射电镜(HRTEM)分别在JEM 2100Plus和JEM2100F上进行,加速电压200kV。X射线衍射(XRD)表征使用Bruker D8Advanced focus衍射仪进行,工作电压40kV,工作电流40mA,使用Ni滤光的CuKα(λ=0.154 nm)射线,扫描速率为5(°)/min,扫描角度范围为10°~80°。X射线光电子能谱(XPS)在Thermo Escalab250仪器上进行,以AlKα为光源(hv=1486.6 eV),扫描100次,以C1s取值284.5 eV作为基准校正。催化剂的BET比表面使用ASAP2460物理吸附仪进行测量,经120℃真空预处理2h的样品在240℃吸附N26h,并在77K下记录样品的N2吸脱附等温线。程序升温还原(TPR)实验在Micromeritics Auto Chem II2920型全自动化学吸附仪上进行,具体操作步骤如下:首先,取60mg催化剂于U形石英管中,然后,将催化剂在120℃、30ml/min的氩气气氛下吹扫干燥1.5 h;最后,在5%H2/Ar(30ml/min)气氛中以5℃/min的升温速率升温至800℃并记录TCD信号。利用红外(FT-IR)表征催化剂的表面基团,催化剂与KBr按照1∶100的比例混合压片,在Thermo Scientific Nicolet iS10上进行测量。催化剂的热重分析在Mettler差热热重测定仪上进行:称取20mg样品在50ml/min流速的N2气氛下从室温加热到800℃,记录DTG曲线。
1.3 催化性能测试
将1mmol HMF、50mg催化剂和10ml DMF在30ml的反应釜内衬中搅拌均匀后装釜,置换三次氧气以排出反应釜内空气,在700r/min的搅拌速率下加热至110℃,恒温反应到一定时间后通过骤冷停止反应,将催化剂过滤后得到反应液。反应液经稀释后使用HPLC(Shimadzu LC-20A)通过RID和PDA检测器定量测量反应产物。实验中所使用的色谱柱为Shodex SUGARSH-1011column(8mm×300mm),使用流动相为0.00 5mol/L的稀H2SO4溶液,流速0.9ml/min,柱温为40℃,进样量5μl。
采用转化率、选择性和表面活性描述催化剂的催化性能,计算公式为:
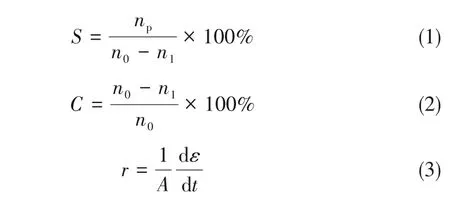
式中,S为选择性,%;C为转化率,%;n0为HMF的起始量,mmol;n1为HMF的剩余量,mmol;np为DFF的生成量,mmol;r为催化表面活性,mmol/(m2·h);A为BET表面积,m2;ε为反应物转化量,mmol;t为反应时间,h。
2 实验结果与讨论
2.1 催化剂的物相和形貌分析
图2为MnO2纳米催化剂的XRD谱图。在水热合成的MnO2-NF、MnO2-NW1、MnO2-NW2和MnO2-NW3样品谱图上均可与隐钾锰矿结构的α-MnO2的标准卡片(JCPDS44-0141)进行精确对应,所观察到位于12.78 °、18.10 °、25.71 °、28.84 °、37.52 °、41.97 °、49.86 °和60.27 °的衍射峰分别对应了α-MnO2的(110)、(200)、(220)、(310)、(211)、(301)、(411)和(521)晶面。商业二氧化锰MnO2-NR具有混晶结构,主要成分为六方锰矿结构的Mn(OOH)2(JCPDS17-0510),同时可观察到少量可与α-MnO2对应的晶相峰。而且,MnO2-NW1、MnO2-NW2和MnO2-NW3的晶相衍射峰整体比MnO2-NR和MnO2-NF更明显和尖锐,这表明后两种催化剂的结晶度较低。在MnO2-NW1、MnO2-NW2和MnO2-NW3三种具有较高结晶度的催化剂中,主衍射峰(310)和(211)的峰高比值存在明显差异。其中,MnO2-NW2的(310)/(211)峰高比最大,表明其表面(310)晶面暴露的比例最高,而MnO2-NW2谱图在37.52 °处的(211)晶面其衍射峰比MnO2-NF、MnO2-NW1、MnO2-NW3的晶相衍射峰的峰强都弱,根据文献[24],氧化锰中Mn3+量减少以及晶格氧量增加有可能降低氧化锰(211)晶面的暴露度,从而减弱谱图在37.52°处的(211)晶面衍射峰。表1中的相应数据表明,相比其他样品MnO2-NW2具有较低的Mn3+含量和较高的晶格氧含量。
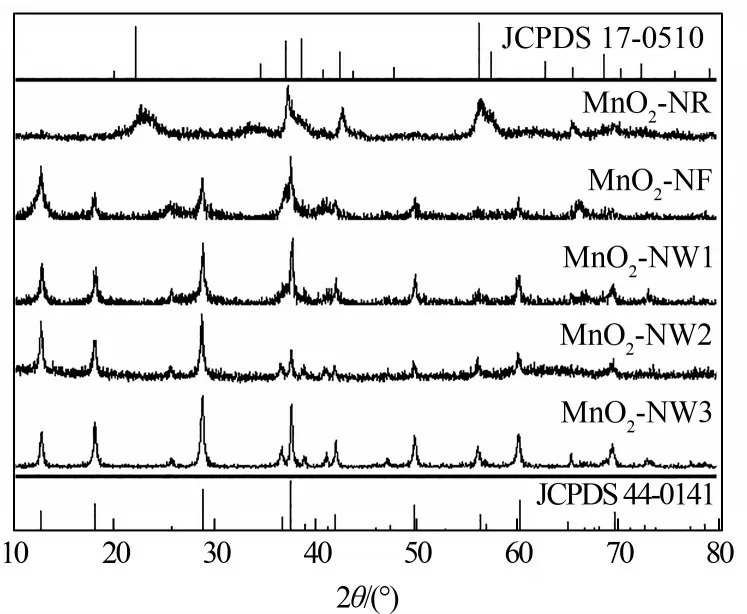
图2 不同二氧化锰催化剂的XRD谱图Fig.2 XRD patterns of different manganese dioxides
图3为MnO2纳米催化剂的微观形貌。从图3(a)可见,在较低水热温度下制备的MnO2-NF(140℃)是由纳米片组装成的花状结构,其HRTEM图可观察到少量间距为0.47 nm的晶格条纹,对应α-MnO2的(200)晶面。图3(b)~(d)中,升高水热温度至170~200℃所制备的MnO2-NW1、MnO2-NW2、MnO2-NW3样品均有线装纳米结构,有文献报道这是由于纳米片为降低表面能发生卷积形成了纳米线[25],在它们的HRTEM图中可观察到的晶格条纹明显增多,其中,间距为0.31 nm和0.24 nm的晶格条纹组分别对应α-MnO2的(310)和(211)晶面。从图3(e)可见,商业二氧化锰MnO2-NR主要由短而均匀的纳米棒组成,间距为0.40 nm的晶格条纹可对应六方锰矿的(101)晶面。从HRTEM图中观察到晶相信息与XRD谱图的分析结果一致。

图3 二氧化锰催化剂的SEM、TEM和HRTEM图Fig.3 SEM,TEM and HRTEM images for MnO2-NF,MnO2-NW1,MnO2-NW2,MnO2-NW3and MnO2-NR respectively
2.2 催化剂活性及表面电子状态比较
图4比较了五种二氧化锰催化剂在HMF氧化反应中的催化性能。在相同反应条件下,具有纳米线结构且结晶度较高的MnO2-NW1、MnO2-NW2和MnO2-NW3催化剂相比结晶度较低的纳米花状MnO2-NF获得了更高的HMF转化率和DFF选择性,商业MnO2-NR的活性最低,其HMF转化率仅为MnO2-NF的1/3。其中,MnO2-NW2具有最优的催化性能,约72%的HMF转化率和99%的DFF选择性。在前文XRD表征中发现MnO2-NW2催化剂表面(310)晶面的暴露比例比其他催化剂高,Zhang等[26]曾报道α-MnO2的(310)晶面比其他晶面具有更多的氧空位和更强的氧气活化能力,这可能是MnO2-NW2获得最高催化活性的原因之一。

图4 不同二氧化锰催化剂在DMF为溶剂下的转化率和选择性[反应条件:1mmol HMF,110°C,0.5 h,0.5 MPa,催化剂(50mg),n(MnO2/HMF)=0.55]Fig.4 Product conversion and selectivity in aerobic oxidation of HMF over different MnO2with DMF[reaction condition:1mmol HMF,110°C,0.5 h,0.5 MPa,catalyst(50mg),n(MnO2/HMF)=0.55]
根据Liu等[18]和Suib等[22]对二氧化锰催化醇类氧化反应的机理研究结果,催化剂中锰的氧化还原状态和晶格氧的含量对催化剂的活性有重要影响。为了进一步明确反应活性中心及其本质影响因素,对四种α-MnO2催化剂进行了XPS和TPR表征,用以分析各催化剂的氧化还原能力。为消除不同催化剂的比表面积差异对催化性能的影响,对各种催化剂的表面活性也进行了测算,结果见表1。
图5(a)~(c)分别为各催化剂的Mn2p、Mn3s和O1s谱 图。图5(a)中 结 合 能 为643.9 、642.4 、640.8 eV的峰分别对应Mn4+、Mn3+和Mn2+物种;图5(c)中结合能在529.9 、531.4 eV附近的峰分别代表晶格氧(Olatt/Oads)和吸附氧(Oads)物种[27]。通过积分各特征峰可计算出各Mn、O物种的比例,计算结果如表1所示,Mn4+/Mn3+和Olatt/Oads的比例大小的顺序均为MnO2-NW2>MnO2-NW3>MnO2-NW1>MnO2-NF,与各催化剂的表面活性顺序一致。表明催化剂的本征活性与Mn4+/Mn3+比例、晶格氧含量密切相关。据Shen等[28]报道,催化剂中锰和氧的不同价态会导致催化剂的电子结构发生相应的变化,Mn4+/Mn3+比例和晶格氧含量都会直接影响催化剂的平均氧化态AOS。各种催化剂的AOS可通过XPS和H2-TPR两种方法进行测算。Mn3s轨道分裂情况对氧化锰的锰价态变化最敏感,因此可以利用图5(b)XPS谱图测算各催化剂的AOS,计算公式为AOS=8.0-0.9 ×Δ(Mn3s),其中,Δ(Mn3s)为Mn3s轨道分裂峰之间的间距[26]。
由图5(d)的H2-TPR曲线可以观察到,所有催化剂的还原峰均由两个重叠的峰组成,这些峰可以归因 于MnO2-Mn2O3(Mn3O4)-MnO的 还 原 过 程[29]。MnO2-NF的第一个还原峰相比于其他催化剂向低温移动,除此之外,其他三个催化剂峰的外形相似,表明它们具有相似的还原能力。催化剂的还原能力也对HMF氧化性能至关重要,有研究表明,高氧迁移率有利于催化剂表面的氧转移,从而提高了催化氧化反应的活性[30-31]。例如Chen等[32-33]研究发现,MnOx和Co或Fe的协同作用能使Mn氧化物的TPR还原峰降低,从而获得更高的氧迁移率和催化氧化活性。
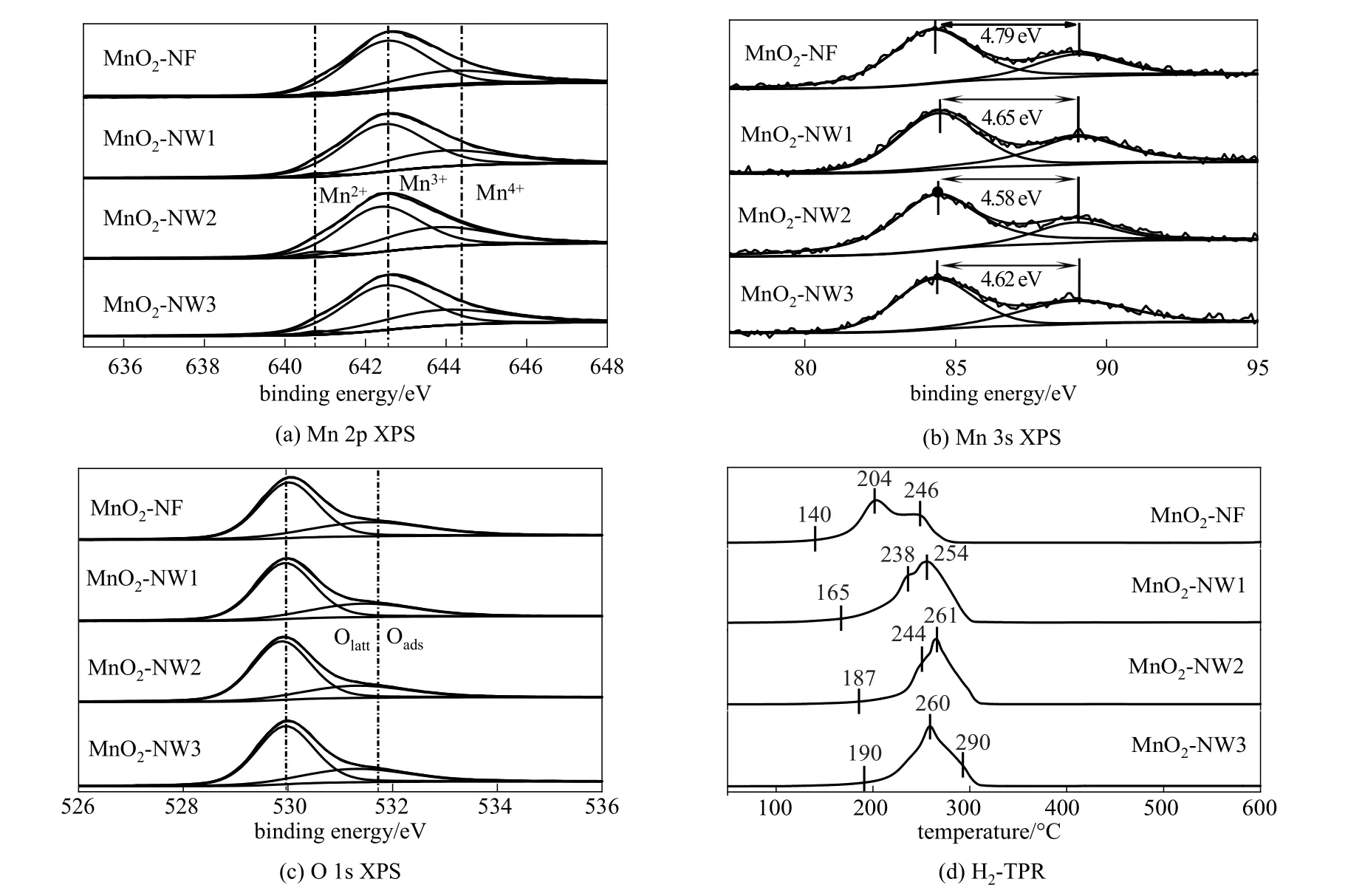
图5 二氧化锰催化剂的表面电子结构表征Fig.5 Characterization of surface electronic structure of manganese dioxide catalyst
此外,以CuO作为内标,通过H2消耗峰的面积也可以计算催化剂的AOS。计算结果均列于表1中,可以发现通过XPS和TPR两种方法求出的催化剂AOS的结果非常接近。催化剂AOS的大小顺序为MnO2-NW2>MnO2-NW3>MnO2-NW1>MnO2-NF,与它们的表面活性顺序一致。
2.3 氧化还原气氛处理的影响
由于催化剂AOS与反应活性之间呈正相关,可预见通过氧化还原气氛处理改变一个催化剂的AOS很可能会影响其反应活性。参照MnO2-NW2的H2-TPR初始还原温度180℃,选取140、180、200℃三个温度对MnO2-NW2在H2气氛中进行40min的 还 原 处 理,分 别 得 到R140、R180和R200三个样品。再将部分R200在300℃的空气气氛中进行1h的氧化处理,得到R200-O300样品。图6显示了这些经过氧化还原气氛处理所得样品的HMF催化氧化活性,HMF转化率变化表明:通过还原气氛处理降低AOS会降低催化活性,而通过氧化气氛处理升高AOS能够提高催化活性。

图6 表面还原氧化处理对催化活性的影响[反应条件:1mmol HMF,110℃,0.5 MPa O2,10ml DMF,1h,n(MnO2/HMF)=0.55]Fig.6 Effect of surface reduction oxidation treatment on catalytic activity[reaction conditions:1mmol HMF,110℃,0.5 MPa O2,10ml DMF,1h,n(MnO2/HMF)=0.55 ]
2.4 催化剂失活和再生
催化剂的稳定性是评价催化剂的重要因素之一,对活性最高的MnO2-NW2催化剂的循环使用稳定性进行了测试。图7(a)展示了每次循环使用前只经过水和乙醇冲洗的催化剂反应结果,可以观察到催化剂存在严重的失活现象,从第一次使用到第五次循环HMF的转化率从79%降低到了50%。将使用后的催化剂进行结构表征,从图8可以观察到,使用后的催化剂相比新鲜催化剂在XRD、FT-IR、XPS谱图和DTG曲线上都发生了变化,其中FT-IR谱图和DTG曲线上均出现了一对新的特征峰,代表反应结束后部分反应底物或产物会吸附到催化剂的表面,而XPS特征峰的偏移及Mn3s轨道分裂峰之间的间距变大则表明使用后催化剂的AOS有所降低。
根据前文的氧化还原气氛处理实验,将使用失活后的催化剂在空气气氛下300℃焙烧1h再进行相同的活性测试,发现HMF转化率可以恢复到75%。空气焙烧法通过把催化剂进行空气焙烧,能够加速Mn3+向Mn4+转变的速度,改变AOS,从而使催化剂活性恢复。图7(b)展示了每次循环使用前都经过空气焙烧再生的催化剂反应结果,催化剂经过6次循环使用后仍能保持>95%的初始活性。以上结果表明氧化气氛处理能使催化剂再生。将再生后的催化剂进行结构表征(图8)可以观察到,其XRD、FT-IR、XPS谱图和DTG曲线基本与新鲜催化剂相同,证明空气焙烧的再生过程能很好地恢复催化剂的表面结构。图9以AOS为横坐标对不同状态的α-MnO2催化剂的表面活性进行关联,验证了AOS与催化活性呈正相关的构效关系。
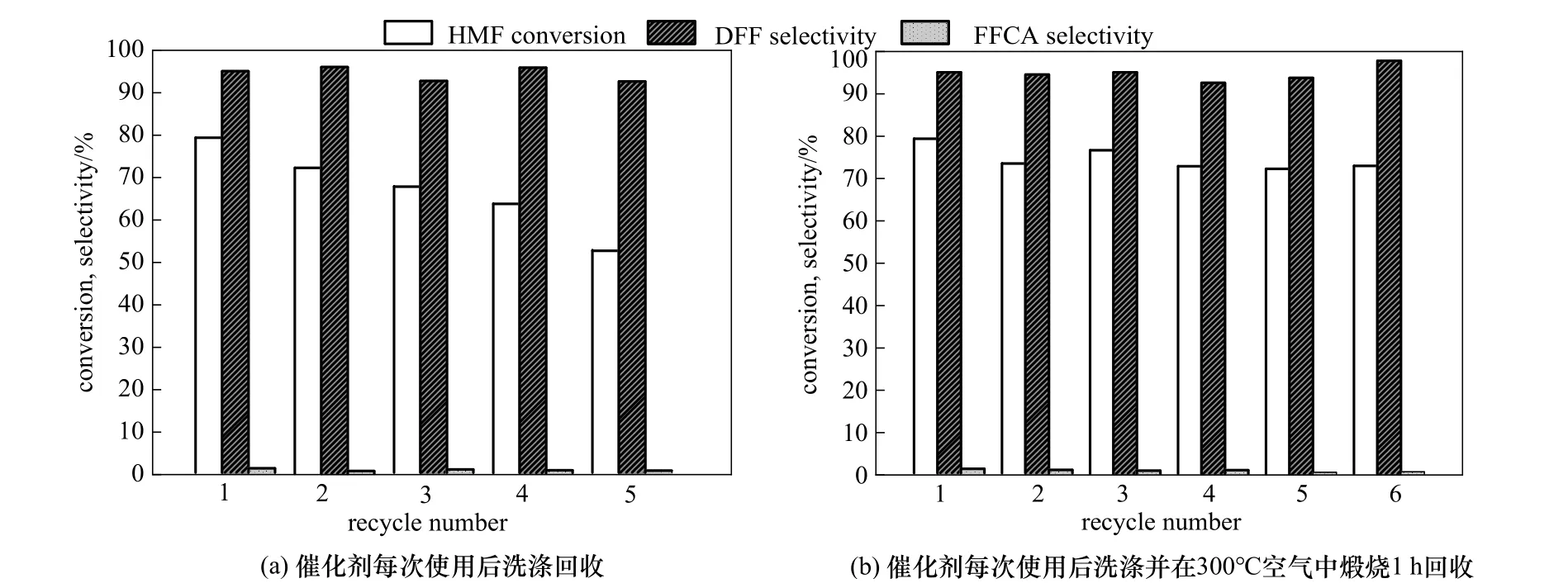
图7 MnO2-NW2催化剂的循环使用性能Fig.7 Recycle performances of MnO2-NW2for the aerobic oxidation of HMF
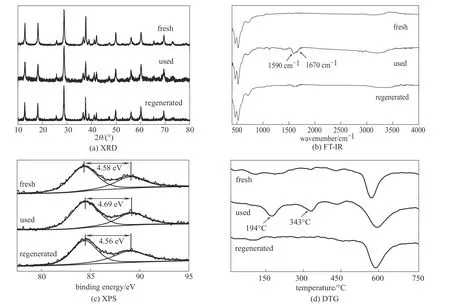
图8 各状态MnO2-NW2催化剂的结构表征Fig.8 Structural characterization of MnO2-NW2catalysts in various states
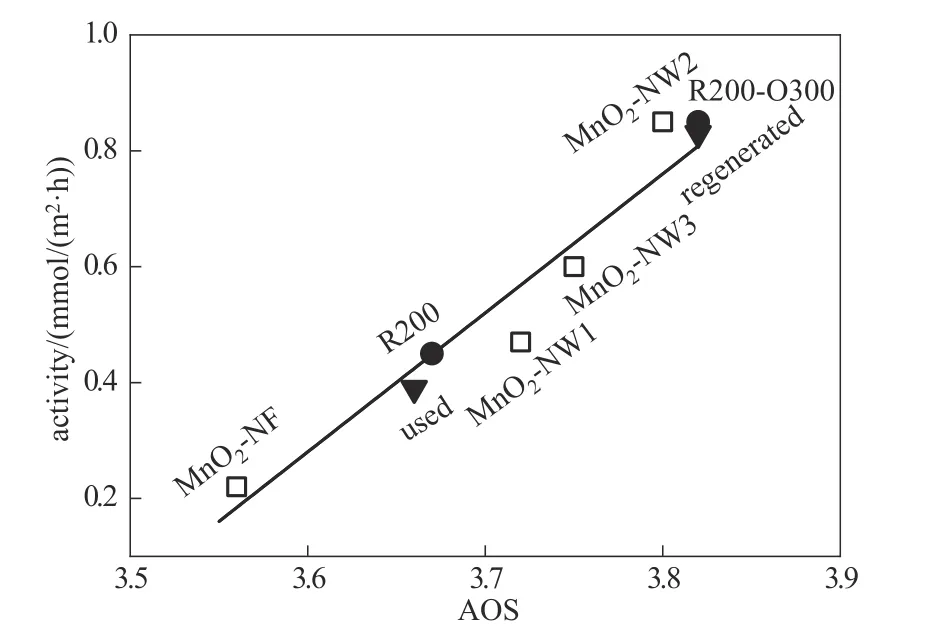
图9 AOS和催化剂活性之间的关系Fig.9 Dependence of AOS and catalytic activity for MnO2 catalysts
3 结 论
本文通过简单的水热法制备了四种具有不同形貌和表面氧化态的二氧化锰催化剂用于HMF选择性氧化制备DFF的反应。研究发现这些催化剂的AOS与催化活性呈正比关系,通过氧化/还原气氛焙烧可以对其表面氧化态进行有效调控,从而调变催化活性。经过优化筛选,使用MnO2-NW2纳米线作催化剂,反应1h可获得79%的HMF转化率和99%的DFF选择性,经过空气焙烧再生,可在6次循环使用后仍保持>95%的初始活性。
猜你喜欢
太原理工大学学报(2022年5期)2022-09-23
农村青少年科学探究(2022年3期)2022-05-13
当代作家(2021年11期)2021-12-17
科学(2020年4期)2020-11-26
中国金属通报(2020年20期)2020-03-27
科学(2020年4期)2020-01-11
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
云南民族大学学报(自然科学版)(2015年4期)2015-11-14
化学教学(2015年1期)2015-03-19
