
石墨相氮化碳光催化剂的研究进展
2021-12-31 08:25:46孙有为胡绍争
石油化工高等学校学报 2021年6期
谢 磊,刘 帅,孙有为,张 健,胡绍争
(1.中国石油东北炼化工程有限公司葫芦岛设计院,辽宁葫芦岛125000;2.辽宁石油化工大学 石油化工学院,辽宁 抚顺113001)
随着社会的不断进步与发展,能源需求方面的挑战和环境污染的问题日益增加。不可再生能源如煤、石油、天然气的过度消耗,造成了人类社会中可利用的资源越来越少,而过度消耗资源带来的环境污染问题也困扰着人类的日常生活[1]。随着工业化的快速发展和人口的持续增长,预计到2050 年,全球的能源消耗将是目前需求量的两倍。解决能源转换和环境污染问题是我国实现可持续发展,实现人民幸福生活,保障国家安全的重中之重[2]。
光催化技术因其在能源和环境应用方面的巨大潜力而受到了科研工作者的广泛关注。1972 年,A.Fujishima 等[3]发现了TiO2电极可以在紫外光照射下分解水,该发现就此拉开了光催化领域的序幕。随着科研人员的不断探索与研究,相继发现了TiO2作为光催化剂可以降解有机污染物[4],半导体粉末如TiO2、ZnO、SiC、GaP、CdS 能够利用光催化技术将CO2还原为各种有机化合物[5]。将光能直接转化为化学能被认为是解决未来能源和环境污染危机的有效途径之一。
在光催化技术中,除了光能作为反应驱动力之外,合适的催化剂也是实现高效光催化的必要条件。一些具有紫外光响应和可见光响应的催化剂如TiO2、ZnO、CdS、Bi2WO6等在光解水产氢、CO2还原、有机污染物降解、有机化合物选择性合成等方面展现出了一定的效果。虽然传统的光催化剂在光催化领域应用广泛,但是带隙能过大,对光能利用率较低,限制了其进一步的发展[6]。石墨相氮化碳(g⁃C3N4)作为一种非金属半导体材料具有制备简单、无毒无害、带隙能可调控、稳定性高等特点,被看作是一种“绿色材料”,广泛用于光解水产氢和光催化降解有机污染物领域。在可见光的照射下,g⁃C3N4可以产生具有氧化能力的光生空穴和具有还原能力的电子,进而参与到相应的氧化还原过程中。但是氮化碳禁带宽度较大,导致其对可见光的吸收能力下降,此外较小的比表面积也严重制约其在光催化领域的应用[7]。 为了更高效地利用g⁃C3N4,科研人员对其进行了一系列的改性。本文对近年来g⁃C3N4的制备合成、改性应用进行了简要的综述,对改性催化剂的反应机理进行了研究与探讨,对其未来发展进行了展望。
1 g⁃C3N4的制备
1.1 g⁃C3N4的制备方法
E.Kroke 等[8]对g⁃C3N4进行密度泛函理论计算(DFT)得到的结果以及L.Pauling 等[9]进行的X 射线单晶衍射实验结果均证明了g⁃C3N4层状结构是由共平面三嗪环或者3⁃s⁃三嗪环单元组成(见图1)。g⁃C3N4自身优异的热稳定性得益于其层间良好的范德华力作用。同时其A⁃B 型堆叠结构降低了层间作用力,增强了电子密度的均匀分布,为g⁃C3N4提供了良好的化学稳定性[10]。

图1 g⁃C3N4的层状结构Fig.1 The layered structure of g⁃C3N4
自然界中g⁃C3N4都不是天然形成的,而是经过人工合成得到的。只需要选取合适的富氮前驱体,例如尿素[11]、氰胺[12]、双氰胺[13]、三聚氰胺[14]、硫 脲[15]等,在适当条件下通过热解法、固相反应法、溶剂热法和电化学沉积法均可以成功制备出g⁃C3N4[16]。在上述方法中,热解法是制备g⁃C3N4的主要方法。该方法通过对反应温度进行调控,对前驱体进行热诱导缩聚,在不同的温度下可以形成三聚氰胺、3⁃s⁃三嗪,最终在520 ℃下形成g⁃C3N4[17]。同时该方法所选用前驱体种类众多,制备过程对反应条件和设备操作要求较低,可以大规模的进行g⁃C3N4的生产。
1.2 不同焙烧温度对制备g⁃C3N4的影响
前驱体类型和制备过程的反应条件影响g⁃C3N4的碳氮原子数比、比表面积、孔隙率、吸收带位置、纳米结构等理化性质[18]。S.C.Yan 等[14]通过对三聚氰胺进行不同温度的焙烧,发现在500、580 ℃下焙烧得到g⁃C3N4的C/N 原子数比分别为0.72 和0.74,均低于理论值的0.750,而这两种催化剂的带隙分别为2.80 eV 和2.75 eV。Z.Mo 等[19]发现焙烧温度的变化会影响到g⁃C3N4的晶形结构、形貌和带隙。随着焙烧温度的升高,g⁃C3N4的形貌会变得更加蓬松,其层状结构厚度会变薄。当焙烧温度在600~650 ℃时,g⁃C3N4表面会出现大小不一的介孔结构。这说明不同焙烧温度会影响前驱体的聚合程度,导致催化剂的形貌有所差异。同时,随着焙烧温度的升高,得到的g⁃C3N4粉末颜色从淡黄色变成了暗橙色。对不同焙烧温度下得到的催化剂进行UV⁃Vis DRS 表征,发现焙烧温度的升高使g⁃C3N4的吸收带由470 nm 红移至570 nm,吸收带的红移意味着催化剂带隙的减小,而带隙的变化影响着催化剂对可见光的吸收程度。D.Hollmann 等[20]通过溶胶⁃凝胶法在不同温度(350~600 ℃)下制备出SG⁃C3N4催化剂,利用电子顺磁共振(EPR)考察了反应温度对催化剂电荷分离和电子转移的影响。结果发现,当焙烧温度由450 ℃升高至600 ℃时,由于其表面捕获的电子数量增多,所制备的催化剂EPR 信号显著增强,这进一步说明了焙烧温度的升高加强了催化剂的聚合程度,增强了催化剂中载流子的分离效果。
1.3 不同前驱体对制备g⁃C3N4的影响
除了氰胺、双氰胺和三聚氰胺可以作为合成g⁃C3N4的前驱体外,以硫脲为前驱体通过热解法也可制备g⁃C3N4。G.G.Zhang 等[15]通过直接焙烧硫脲得到了g⁃C3N4,经过相关表征发现前驱体中硫的存在提高了g⁃C3N4层间结构的连通性和填充性,增强了硫脲的聚合程度,从而增强了芳香层中电子的离域作用,提高了催化剂的性能。Y.W.Zhang 等[21]利用尿素受热可以产生气体的特点,在550 ℃下焙烧3 h得到了具有介孔结构的皱褶薄片g⁃C3N4。在热处理过程中,尿素在低温(<200 ℃)下产生的NH3和高温下产生的CO2气体起到造孔的作用。表面介孔结构的存在可以增大g⁃C3N4的比表面积,有利于光催化作用的增强。D.J.Martin 等[22]分别对尿素、硫脲、双氰胺焙烧所得到的g⁃C3N4进行形貌表征,发现由于多孔结构和片状结构的存在,得到的样品比表面积和孔径明显增大。而对于硫脲和双氰胺而言,热处理得到的g⁃C3N4均没有呈现出孔结构特征。
1.4 不同热聚合时间对制备g⁃C3N4的影响
在催化剂制备的过程中,热聚合时间的不同对催化剂形貌也会产生一定影响。F.Dong 等[23]考察了在550 ℃,不同热聚合时间对尿素生成g⁃C3N4的影响。发现当热解时间从0 延长到240 min 时,g⁃C3N4的比表面积显著增大,由31 m2/g 增加到288 m2/g,这是因为聚合时间的延长增加了g⁃C3N4的聚合程度,提高了孔隙率,导致催化剂层厚度的降低。通过TEM 表征发现,当反应条件为550 ℃,聚合时间为0 时,所制备的g⁃C3N4由较大且连续弯曲的层面构成。当聚合时间变为60 min 时,g⁃C3N4形成了尺寸减小和厚度变薄的层状形貌。而当聚合时间延长至240 min 时,观察到g⁃C3N4呈现出疏松、柔软的丝状纳米结构,上述内容均可以说明不同的热处理时间会造成g⁃C3N4形貌的差异。
1.5 不同反应气体对制备g⁃C3N4的影响
g⁃C3N4的合成除了会受到前驱体类型、反应温度和反应时间的影响外,其独特性质和化学结构也会受到反应过程中气体氛围的强烈影响,不同的气体氛围会诱导g⁃C3N4产生无序结构、缺陷和碳氮空位。通常情况下,前驱体均是在空气氛围下热聚合形成g⁃C3N4。P.Niu 等[24]在H2氛围下制备了含有氮缺陷的g⁃C3N4。H2可以从催化剂表面扩散到其内部空间中,导致催化剂的层状结构中出现空位。除了上述讨论以外,表1 总结了在不同前驱体和反应条件下制备得到的g⁃C3N4比表面积和带隙数据。由表1 可见,适当调整反应条件和前驱体可以改变g⁃C3N4的带隙结构和比表面积,这对改善g⁃C3N4的光催化性能起到了一定作用。
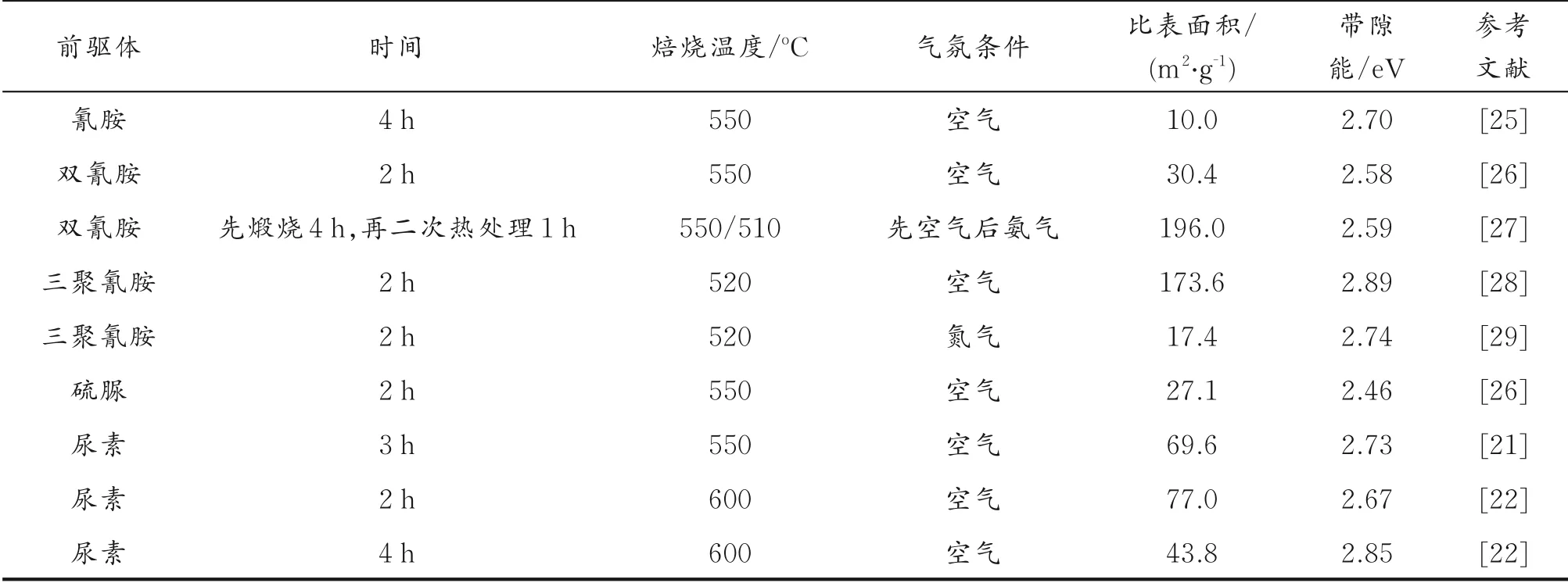
表1 不同前驱体, 反应条件对g⁃C3N4比表面积和带隙的影响Table 1 The influence of different precursors and reaction conditions on the specific surface area and band gap of g⁃C3N4
2 g⁃C3N4的改性
2.1 形貌结构改性
虽然g⁃C3N4在光催化领域应用广泛,但是g⁃C3N4本身存在的一些问题制约了其光催化性能。例如颗粒粒径较大会导致g⁃C3N4的比表面积较小(10 m2/g),造成催化剂表面活性位点数量很低[30]。此外当g⁃C3N4被光激发后,产生的光生电子空穴对容易发生复合,导致量子效率低下。同时,其自身对可见光的响应范围比较窄,也会影响g⁃C3N4对太阳光的利用效率。以上缺陷使得g⁃C3N4在光催化领域的发展受到限制,不能很好地应用到实际生活中。为此,科研人员开展了大量的研究工作,包括对g⁃C3N4进行形貌改变、引入缺陷、掺杂元素、沉积贵金属、与g⁃C3N4复合构成异质结材料等。
通过对g⁃C3N4进行形貌、结构、尺寸上的优化,增大其比表面积;通过增加其表面活性位点的数量来提高催化剂与反应物分子的接触程度,从而提升传质效率,以达到较高的反应活性。不同形貌结构的g⁃C3N4会呈现出不同的比表面积和孔容孔径,对普通g⁃C3N4进行形貌和结构的改性,例如制备纳米结构,形成介孔结构可以增大催化剂的比表面积,增加表面活性位点数量,从而提高催化性能。此外,将缺陷引入到g⁃C3N4中,通过降低带隙可以提高催化剂对可见光的响应能力。同时缺陷的存在也可以抑制光生电子空穴对的复合,提高量子效率,强化催化剂性能。P.Niu 等[24]利用高温烧结法将氮缺陷引入到g⁃C3N4中,相比纯g⁃C3N4,具有氮缺陷的g⁃C3N4带隙能降低了0.75 eV,带隙能的降低可以提高催化剂对可见光的响应能力。M.Wu 等[31]在不添加模板剂的情况下,通过水热共聚法制备了具有高比表面积和氮缺陷的三维纳米多孔石墨氮化碳,在光催化产氢的实验中,其产氢速率为92.57 μmol/h,是普通g⁃C3N4的20.3 倍。催化剂中氮缺陷与纳米结构的协同作用使催化剂具有更宽的可见光响应范围,同时较大的比表面积(226.19 m2/g)使催化剂表面暴露出更多的催化活性位点,活性位点的增多使更多的H+在催化剂表面参与反应。在可见光激发下,催化剂产生光生电子⁃空穴对,大量的H+与催化剂导带上的电子反应产生了H2,其产氢机理如图2 所示。

图2 三维纳米多孔石墨氮化碳的光催化产氢机理Fig.2 Photocatalytic hydrogen production mechanism diagram of three⁃dimensional nanoporous graphite carbon nitride
C.B.Chen 等[32]利用热聚合过程中氰胺前驱体的不完全缩聚,采用硬模板剂合成法制备了具有较高催化活性的介孔氮化碳纳米球光催化剂(MCNS)。相比于块状形貌和纳米片形貌的g⁃C3N4,MCNS 展现出更大的比表面积和更丰富的氰基结构。其较大的比表面积和大量的活性位点有利于产生更多的光生电子⁃空穴对。而氰基也扮演着“缺陷”的作用,促进光生电子空穴对的分离。在光催化降解双酚A 的过程中,光激发后MCNS 产生电子和空穴,价带上的空穴可以与催化剂表面的双酚A 发生氧化反应,而电子可以与催化剂表面的吸附氧发生还原反应生产超氧自由基不仅可以直接氧化双酚A,也可以进一步反应生成羟基自由基(·OH),进而共同参与双酚A 的降解(见图3)。MCNS 独特的纳米球结构、高比表面积和丰富的氰基使其在光催化降解双酚A 反应中具有优越的活性。其一级动力学反应常数为0.094 min-1,是普通g⁃C3N4的14.7 倍。

图3 MCNS 光催化降解双酚A 机理Fig.3 The mechanism of MCNS photocatalytic degradation of bisphenol A
2.2 掺杂改性
掺杂改性可以调节催化剂本身的电子能带结构,有效抑制光生电子空穴对复合,提高催化剂的光催化能力。B.Yue 等[33]利用Zn2+更容易配对g⁃C3N4的 特 点,将g⁃C3N4与ZnCl2在HCl 的 作 用 下 制备出Zn/g⁃C3N4催化剂,一方面可以让掺杂的Zn2+作为电子捕获剂和产氢反应的活性中心。另一方面可以使掺杂的Zn 通过Zn-N 键连接在氮化碳上,由于化学键的存在,Zn2+不需要Cl-来平衡电荷,所以Zn/g⁃C3N4中不会出现杂质相,当Zn 的掺杂量合适时,相比于纯g⁃C3N4,Zn/g⁃C3N4的产氢速率是它的10 倍。J.N.Zhu 等[34]以三聚氰胺和CuCl2·2H2O 为原料,将二者合成为三聚氰胺模板化结晶氯化铜[H2mela]2[CuCl5]Cl,之后通过高温裂解制备掺铜催化剂Cu⁃g⁃C3N4。对Cu⁃g⁃C3N4进行XPS、FT⁃IR 表征,发现其内部含有大量的Cu-Nx配位键,具有较强反应活性的Cu-Nx键在pH 为中性的条件下,可以与H2O2反应生成·OH。同时在光照条件下,价带上的空穴可以直接氧化有机污染物,导带上的电子可以还原氧气,形成·O-2参与到污染物的降解中,其降解机理如图4 所示。
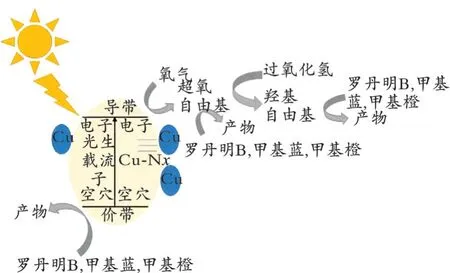
图4 Cu⁃g⁃C3N4光催化降解污染物机理Fig.4 Cu⁃g⁃C3N4 photocatalytic degradation mechanism of pollutants
H.H.Ou 等[35]采用两步连续热处理法,成功地制备了含硒的多孔纳米板氮化碳材料(m⁃CNNSs)。在可见光照射下,产氢速率达到了130 μmol/L,是普通氮化碳的7 倍。光催化性能的提高主要受益于较大的比表面积,多孔纳米结构的存在加速了光生载流子的分离,抑制了光生电子空穴对的复合。此外硒的掺杂使催化剂带隙减小,扩大了催化剂对可见光的吸收范围。T.Huang 等[36]以双氰胺作为唯一原料,采用低温水热两步法和后续焙烧成功制备了具有氮空位和氧掺杂的多孔棱镜状石墨相氮化碳。由于具有孔隙结构层次分明的多孔棱镜形貌,催化剂的比表面积高达220.16 m2/g。较大的比表面积和特殊的形貌可以暴露更多的氮缺陷活性位点,在可见光照射下,催化剂中的掺杂氧和氮空位可以使导带上的光生电子快速转移至氮缺陷上,与活性氮反应生成氨(其固氮反应机理,见图5),从而使催化剂具有优异的光催化固氮性能(118.8 mg/(L⋅h⋅g))。

图5 多孔棱镜状石墨相氮化碳的光催化固氮反应的机理Fig.5 Mechanism of photocatalytic nitrogen fixation of porous prismatic graphite carbon nitride
2.3 贵金属负载
将贵金属如Ag、Au、Pt、Pd 等负载到g⁃C3N4上之后,可以改变催化剂中的电子分布,从而改变催化剂的表面性质。负载的贵金属与g⁃C3N4之间形成空间电荷层,在受到光激发后,催化剂表面的光生电子迁移至贵金属表面,抑制了光生电子空穴对的复合,增强了催化剂的催化能力。S.W.Cao 等[37]采用胶体吸附⁃沉积法,在石墨相氮化碳(g⁃C3N4)上分别负载了立方、八面体和球形铂(Pt)纳米颗粒。由于Pt 纳米颗粒的不同形貌,其可见光驱动催化活性顺序为:立方铂<八面体铂<球形铂。这是因为Pt 纳米颗粒不同的暴露面所具有的表面原子结构不同,导致了催化剂在光催化反应中活性位点和吸附能的差异。
F.L.Wang 等[38]通过简单的共聚合方法成功地在超薄g⁃C3N4上负载了Ag 单原子(AgTCM/UCN),并将其作为可见光光催化剂,考察了在过硫酸盐存在的条件下降解磺胺甲嘧啶的性能。Ag 离子的存在可以将长波长光变为短波长光,提高了催化剂对光能的吸收利用。由于银离子的存在和硫酸盐产生的有效电荷分离效应,一方面抑制了光生电子空穴对的复合,另一方面可以使光生电子更有效地与过硫酸盐生成·SO-4,与氧气生成·O-2。进一步发生反应可以生成·OH,而多种活性自由基的共同作用可以更好地将磺胺甲嘧啶氧化,以达到理想的降解效果。
2.4 半导体构建异质结
通过与半导体复合形成异质结材料,可以很好地改善g⁃C3N4催化活性。当g⁃C3N4与具有不同能带结构的半导体复合后,形成的异质结材料会具有全新的能带结构。同时二者所拥有的不同价带电位和导带电位可以使复合材料能级发生交叠,在异质结材料的界面处产生能带弯曲,在空间上达到光生电子空穴对分离的目的,从而提高催化剂的催化性能[39]。同时,拥有交错能带结构的异质结催化剂在光照条件下,光生电子从各自的价带跃迁到导带上,价带上保留了光生空穴。由于价带位置与导带位置的不同,导致电子会从高电位导带转移到低电位导带上,而在高电位价带上的空穴则会迁移至低电位价带上,这有利于分离电子和空穴,强化光催化活性。电子和空穴的有效分离可以使更多的空穴参与到相应的氧化反应及光生电子与O2形成的反应或者参与到相关还原反应中。
B.Chong 等[40]采用简单的水热共沉积法将多孔管状g⁃C3N4与CdS 纳米颗粒复合成异质结光催化剂。该催化剂中,多孔结构使g⁃C3N4具有更大的比表面积,可以产生更多的反应活性位点。同时,管状g⁃C3N4与CdS 纳米颗粒之间形成的内置电场也可以加快光生电子和空穴的分离,优化光生载流子的传输性能,所有这些协同作用都有助于催化剂光催化活性的提高。光催化析氢实验可以发现,T⁃CN/CdS 的产氢速率高达71.6 μmol/h,是普通g⁃C3N4产氢速率的16.3 倍。在可见光的照射下,CdS与T⁃CN 均产生了光生电子空穴对,由于二者价带值和导带值的不同,导致了光生空穴聚集在T⁃CN的价带上,而光生电子聚集在CdS 导带上。这一方面很好地抑制了电子空穴对的复合,另一方面可以使H+更好地与光生电子发生反应生成H2。
G.D.Fan 等[41]通过化学沉积法合成了具有p⁃n异质结结构的Ag2O/g⁃C3N4催化剂。通过对催化剂进行活性考察实验,发现当Ag2O 的负载量为14.3%时,反应6 h 后催化剂对微胞藻属绿脓杆菌的消除率达到了99.94%。同时该催化剂具有良好的光稳定性和可重复利用性,这为处理富营养化水体中的蓝藻水华问题提供了很好的发展方向。刘帅等[42]采用浸渍法制备了WO3/g⁃C3N4异质结催化剂,由于WO3和g⁃C3N4存在不同的价带值和导带值,所以电子会从g⁃C3N4导带迁移至WO3导带上,进而还原O2生成·O-2。而空穴则会从WO3价带迁移至g⁃C3N4价带上,从而与H2O 反应生成强氧化性的·OH,而剩余部分的空穴则会直接参与到氧化反应中。在·、·OH 和空穴 对二苯并噻吩(DBT)的共同氧化作用下,该催化剂反应180 min 对DBT 的转化率为72.9%。
臧一鹏[43]通过简单的化学方法制备了SnO2/g⁃C3N4催化剂,导带值和价带值的不同有利于二者形成异质结,促进光生电子空穴对的分离,具体过程为光激发产生电子空穴对后,两种半导体材料的电子均迁移至各自导带上,空穴留在各自价带上。由于g⁃C3N4导带的电位值较高,所以其导带上的电子迁移至SnO2导带上。处于更高电位SnO2价带上的空穴也会转移至g⁃C3N4的价带上,很好地抑制了电子空穴对的复合。同时,SnO2/g⁃C3N4催化剂的导带值低于H+/H2的氧化还原电位(0 eV),所以在光催化产氢实验中,具有还原能力的电子可以将H+还原为H2(产氢机理见图6)。
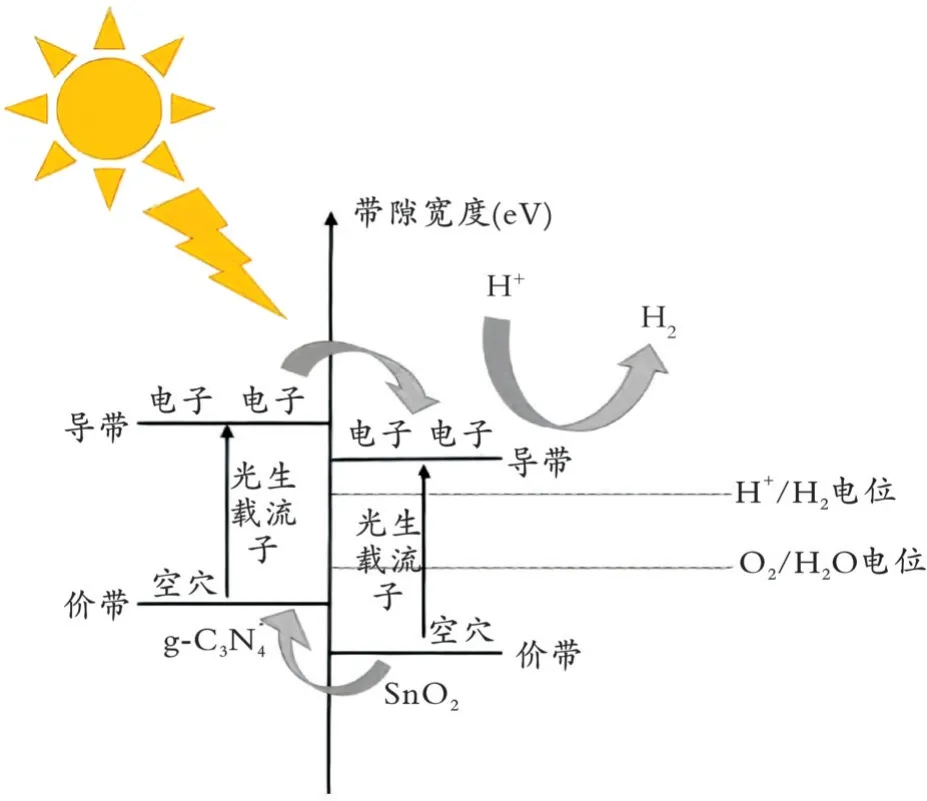
图6 SnO2/g⁃C3N4的光催化产氢机理Fig.6 SnO2/g⁃C3N4 photocatalytic hydrogen production mechanism
2.5 其他改性
J.H.Xi 等[44]通过简单的化学反应和低温焙烧处理,在二维超薄g⁃C3N4纳米片上成功负载了零维赤铁矿量子点。该催化剂利用其较大的比表面积、较多的活性位点和较强的界面耦合作用,提高了电荷传输效率,抑制了光生电子空穴对的复合。此外,由于电荷迁移速率的提高,导致Fe2+/Fe3+的持续快速转化,上述优化作用在光催化降解4⁃硝基酚的实验中得到了很好的体现。Q.Jin 等[45]将构树树叶制备的生物炭,并加入g⁃C3N4粉末与海藻酸钠反应,制备出具有光催化活性的生物炭偶联g⁃C3N4微球(SABC)。在可见光照射下,SABC 产生了大量的光生电子⁃空穴对,生物炭的存在加速了电子的转移,促进了电子与空穴的分离程度,使更多的电子参与到Cr(VI)还原为Cr(Ⅲ)的反应中。同时生物炭较强的吸附能力也加快了反应物分子的传质,提高了反应速率,在光催化还原Cr(VI)反应中表现出良好性能,为水净化领域的实际应用提供了新的发展方向。
D.N.Liu 等[46]采用简单的溶剂热法制备了含有表面氧空位(OVs)的溴化氧铋(BiOBr),并与g⁃C3N4结合生成催化剂CN⁃BiOBr⁃OV,用于光催化氧化NO 和还原CO2。二者形成的异质结可以有效转移光生载流子,同时催化剂表面的氧空位一方面可以对光生电子进行捕获,抑制电子空穴对的复合;另一方面可以使电子与氧气产生更多的超氧自由基用于光催化氧化NO,而具有强氧化性的空穴也可以起到氧化NO 的作用。在异质结和氧空位的协同作用下,CN⁃BiOBr⁃OV 对NO 的去除率达到了63%,而对CO2转化为其他碳质产物的选择性达到了96%。
3 结 论
石墨相氮化碳(g⁃C3N4)作为一种新兴的“绿色”材料,因其原料来源广、廉价、制备简单、稳定性高而成为光催化领域中很有潜力的研究对象。但是,其较低的比表面积会影响到活性位点数量。同时对可见光的吸收效率不高以及光生电子空穴对容易复合的缺点,降低了其光催化活性,限制了g⁃C3N4的进一步应用。尽管科研工作者开发了大量改性方法来克服上述缺陷,但是g⁃C3N4基催化剂依旧存在着可见光利用不足,电子迁移速率低,表面活性位点数量少等缺点。因此在今后的工作与研究中应着重以下几方面:
(1)考察改性g⁃C3N4催化剂活性实验的同时,利用相关理论进一步研究其反应过程变化和催化机理。在获得更加系统完善的机理信息后,对g⁃C3N4进行更高效的改性处理。
(2)g⁃C3N4及其改性催化剂的应用不应局限于光催化产氢和光催化降解污染物的研究中。应加大g⁃C3N4催化剂在其他领域上的探索与研究,例如CO、NO 等气体的氧化反应以及对CO2进行多电子还原生产燃料(CO、CH4、CH3OH)等反应。
(3)面对催化剂改性过程中工艺繁琐、操作复杂、对环境不友好等问题,寻找高效简便和绿色环保的改性方法十分重要。在降低资源损耗,提高催化剂性能的同时,应早日实现g⁃C3N4基催化剂在生产生活中的大规模应用。
猜你喜欢
应用数学和力学(2024年1期)2024-01-29 08:32:46
车用发动机(2021年5期)2021-10-31 05:48:38
装备维修技术(2021年36期)2021-10-25 13:21:04
弹箭与制导学报(2021年3期)2021-07-30 02:56:52
电源技术(2021年7期)2021-07-29 08:35:24
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20 06:33:26
网印工业(2019年4期)2019-05-21 06:41:58
重型机械(2019年2期)2019-04-28 11:52:04
物理化学学报(2017年3期)2017-03-11 00:25:30
昭通学院学报(2016年5期)2016-02-24 10:51:12
