
细胞焦亡机制及其在多种肾脏病中的作用
2021-12-26 01:58孙晓怡姜玉勤唐余燕刘何晶魏明刚
中国医药导报 2021年33期
孙晓怡 姜玉勤 唐余燕 刘何晶 陆 迅 魏明刚
1.苏州大学附属第一医院肾内科,江苏苏州 215006;2.苏州大学附属第一医院中医科,江苏苏州 215006;3.江苏省苏州市立医院北区中医科,江苏苏州 215000
细胞焦亡的主要特征为完整的细胞膜结构破坏、细胞肿胀及细胞质内容物的释放[1]。根据细胞焦亡的发生过程可将其分为两类:经典途径细胞焦亡及非经典途径的细胞焦亡。在多种原因所致的肾脏疾病中炎症反应均可发挥作用。近年研究显示,以炎症小体为核心介导的无菌性炎症反应细胞焦亡为肾脏疾病进展的重要原因[2-5]。因此根据细胞焦亡的特点,采取针对炎症反应的干预措施将会在多种肾脏病的防治中提供优异的作用效果,为临床诊疗带来新的靶点。
1 细胞焦亡产生的机制
1.1 经典途径细胞焦亡
经典途径细胞焦亡为依赖caspase-1 的细胞焦亡,其起始原件为炎症小体,炎症小体主要包括模式识别受体(pattern recongnition receptor,PRRs),前活化半胱天冬酶-1(caspase-1)及凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing,ASC)组成[6]。PRRs 作为炎症小体的感受器,有多种表达形式,在细胞焦亡过程中起主要参与作用的为NOD 样受体(NOD-like receptors,NLRs)[7],NLRs 包括NLRP1、NLRP2、NLRP3 和NLRC4 等,其具有共同分子结构特点:位于中心的核苷酸结合和寡聚化结构域(nucleotide-binding oligomerization domain,NOD),可以发生自身的寡聚;羧基末端亮氨酸重复区(leucinerichrepeat,LRR)发挥识别配体及自我调节的功能;N 末端有可变结构域等[8]。NLRs 可以识别外源性微生物携带的病原相关分子模式(pathogen associated molecular patterns,PAMPs)或内源性细胞损伤释放的相关分子模式(danger-associated molecular pattern,DAMPs),并激活下游促炎症信号级联通路,引起机体的防御或炎症反应[9-13]。
当PRRs 识别外源性信号后作用于NLRs,NLRs通过LRR 进行信号的接受及传递,后通过ACS 信号识别及传递,使NOD 结构参与到炎症小体组装调节过程中,从而间接募集caspase-1 前体激活caspase-1,并进一步作用于下级效应分子gasdermin 家族蛋白D(GSDMD)[14]。GSDMD 为半胱氨酸天冬酶切割的底物,一般情况下保持自抑制构象,其C 末端结构域掩盖了N 末端坏死域中的脂质结合部分,自抑制现象的解除需要外部作用消除C 末端抑制结构域从而暴露出N 末端坏死域后才可进行[15]。在炎症小体的激活过程中,这种C 末端抑制结构域的解除过程即是由炎症性胱天蛋白酶的水解作用实现[16]。N 端坏死域暴露后即可转移至质膜并聚集形成跨膜通道引起细胞肿胀裂解,细胞内容物及炎症因子释放,最终导致细胞焦亡。
1.2 非经典途径细胞焦亡
研究发现除了由caspase-1 介导的经典途径细胞焦亡外,由人源性caspase-4/5 及鼠源性caspase-11 可介导的细胞焦亡途径称为非经典途径细胞焦亡[17]。非经典途径细胞焦亡是细胞焦亡概念的进一步延伸,Kayagaki 研究团队使用霍乱毒素B、大肠埃希菌、啮齿类念珠菌和霍乱弧菌诱导小鼠炎症反应,在caspase-11 基因缺乏的小鼠中细菌所触发的白细胞介素-1α(interleukin-1α,IL-1α)和高迁移率族蛋白-1的释放明显抑制,提示caspase-11 为这一途径的关键因子,此途径可以诱发IL-18 及IL-1β 大量产生从而引起细胞焦亡[18-19]。
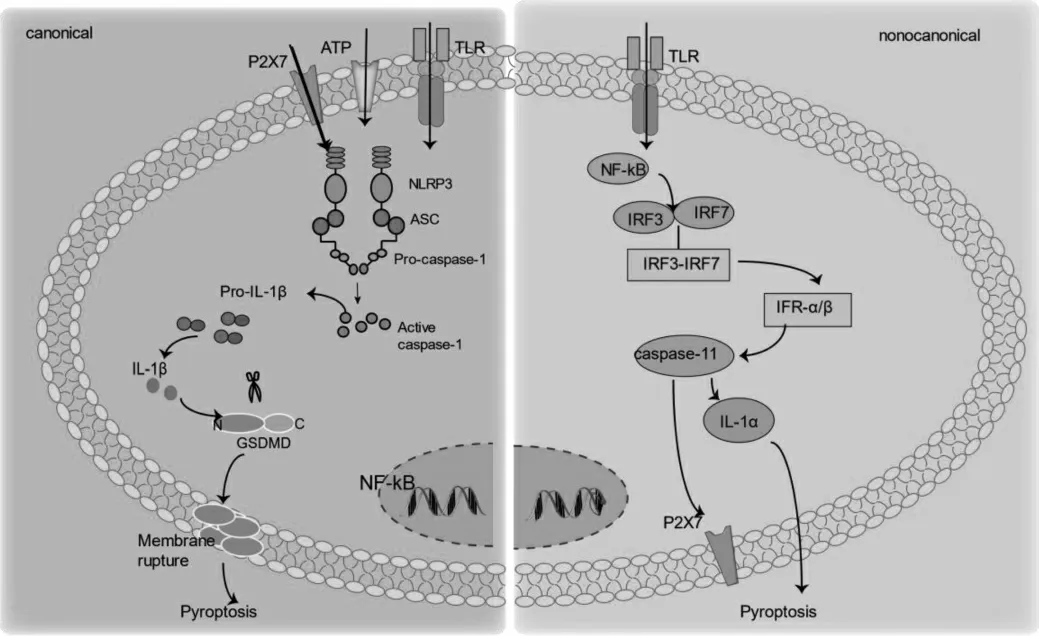
图1 细胞焦亡机制图
2 细胞焦亡机制及其在多种急慢性肾脏病中的作用
2.1 肾脏结石类肾病
肾脏结石类肾病是指尿草酸钙(calcium oxalate,CaOx)负荷过大超过肾脏肾小球滤过和分泌功能,从而导致CaOx 晶体在肾脏中沉积形成肾脏结石或引起弥漫性肾脏钙化[20]。通过分别建立NLRP3 缺乏,ASC缺乏和caspase-1缺乏的小鼠模型,且使用CaOx 晶体与NLRP3 激动剂ATP 进行对照实验,发现CaOx 晶体是通过NLRP3/ASC/caspase-1 途径从而诱导IL-1β 表达,且这一过程需要使用细胞预刺激剂脂多糖(lipopolysaccharide,LPS),提示LPS 预刺激是诱导pro-IL-1β 表达的先决条件。在非经典途径细胞焦亡中,LPS 亦为发生该机制的必要条件,因此或许在草酸盐结晶所致的肾脏病中也会有非经典途径细胞焦亡的参与,对实验中caspase-4/5/11 的检测有助于进一步研究[21]。
2.2 急性肾损伤(acute kidney injury,AKI)
AKI 是临床常见的由各种原因导致的肾功能短时间内快速减退的一种临床综合征,可以由血容量减少、缺血再灌注、造影剂使用及药物原因等多种情况引起,而在以这些因素为始动刺激引起AKI 的过程中,也有细胞焦亡的密切参与。缺血再灌注肾损伤产可通过Toll 样受体降解及其下游所致核因子-κB(nuclear factor κB,NF-κB)炎症及细胞焦亡免疫反应参与AKI的发生过程[22-23]。在使用造影剂主要成分碘丙啶(propidium iodide,IOP)诱导AKI 实验中,可以检测到体内外caspase-1、IL-1β 和IL-18 水平均有显著升高,同时伴有LDH、NLRP3、ASC 和GSDMD 产生,均可提示IOP 可通过细胞焦亡诱导AKI[24]。而在常见的抗肿瘤药物的使用过程中,AKI 作为药物使用的副作用也不罕见,细胞焦亡同样参与其中。在顺铂诱导的小鼠AKI 过程中,肾脏组织及肾小管细胞中均检测到GSDMD 片段的产生,且使用顺铂处理GSDMD 基因缺失小鼠,小鼠发生AKI 及相关炎症反应均较前有所减轻,而活跃或增加GSDMD 片段的操作则可起到相反的作用[25]。
2.3 糖尿病肾病(diabetic nephropathy,DN)
DN 为多种致病因子所致的代谢性疾病,其为一种以糖尿病为基础疾病的微血管并发症。在DN 发病过程中,由炎症小体NLRP3、caspase-1、GSDMD 介导的经典途径细胞焦亡发挥重要作用[26]。研究显示,在肥胖条件下模拟成年小鼠2 型糖尿病模型,内源性大麻素作为外周血CB1 受体激活剂可以通过激活CB1受体并且引起胰岛β 细胞的衰竭,而在NLRP3 缺失的小鼠模型中,β 细胞衰竭现象未明显观察到[27]。另外,在NLRP3 炎症小体缺乏的野生型小鼠中反复低剂量的予以四氧嘧啶可抵抗缺氧及氧化应激所致的细胞损伤,这与胰岛β 细胞衰竭减少及巨噬细胞浸润减少相关,而NLRP3 在这一实验反应过程中扮演着中间介质的角色,NLRP3 缺乏降低了四氧嘧啶诱导的糖尿病患者的β 细胞丢失和胰岛炎症[26,28]。因此,对于这一级联反应进行更为详尽地探索并在此基础上进行相应的干预性措施,可能对DN 的诊疗具有重大的意义。
2.4 肾脏纤维化
肾脏纤维化是慢性肾脏病不断发展至终末期的关键机制,肾组织内炎症因子与抗炎因子之间的失衡,引起组织内过度代谢反应及组织损伤,纤维组织不断增生的同时伴随肾脏固有细胞转化,不断加重肾脏纤维化的进展[29]。研究显示,NLRP3 缺失对于介导线粒体调解肾脏纤维化具有保护作用[30-31]。微生物分子或细胞内危险因子作用于肾组织时,可检测到肾组织中NLRP3、pro-IL-1β 和pro-IL-18 的表达,这可能由NF-κB 通路介导[32],这一过程中伴有活性氧产生,再次作用于NLRP3 引起细胞焦亡,研究显示,TGF-β/Smad 可激活NLRP3,而NLRP3 又可进一步作用于Smad磷酸化过程导致进一步的肾脏纤维化[33],在这一过程中,细胞焦亡过程与肾脏纤维化机制相互作用形成加重细胞损伤的恶性循环[34-35]。
综上所述,细胞焦亡机制为细胞程序性死亡的形式之一,细胞焦亡的发病机制包括经典途径细胞焦亡及非经典途径细胞焦亡,而NLRs、caspase-1、IL-1 及GSDMD 为细胞焦亡通路的关键因子,在多种肾脏病发病及进展过程中均有所参与。在对多种肾脏病中细胞焦亡机制的研究可知氧化应激所致活性氧(reactive oxygen species,ROS)产生增加、炎症小体激活及炎症因子的释放均有较大作用,在针对ROS 产生增加或减少炎症小体的激活对细胞焦亡的进展可起到至关重要的作用,而基于细胞焦亡机制进行更加深入地探索并寻找可能延缓疾病进展的关键环节对于明确肾脏病的发病机制,并且对于多种肾脏病的诊疗亦大有裨益。
猜你喜欢
中老年保健(2022年3期)2022-08-24
世界科学技术-中医药现代化(2022年3期)2022-08-22
医学综述(2022年7期)2022-04-19
现代临床医学(2021年6期)2021-11-20
世界科学技术-中医药现代化(2021年10期)2021-03-02
昆明医科大学学报(2021年1期)2021-02-07
昆明医科大学学报(2020年12期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国生殖健康(2019年10期)2019-01-07
特别健康(2018年9期)2018-09-26
