
利多卡因对大鼠蛛网膜下腔出血后迟发性脑血管痉挛的影响及脑保护作用机制研究
2021-12-24 02:59:46苏飞陈博文李向男刘佳琦陈扬
疑难病杂志 2021年12期
苏飞,陈博文,李向男,刘佳琦,陈扬
蛛网膜下腔出血(subarachnoid hemorrhage,SAH)是一种临床较为常见的脑血管意外,约占急性脑卒中发病率的10%,多由动脉瘤破裂、脑血管畸形、血管炎等引起[1]。脑血管痉挛(cerebral vasospasm,CVS)是动脉瘤破裂后SAH的常见并发症之一,临床主要表现为意识障碍和神经功能受损,是导致SAH患者死亡和致残的重要原因[2]。SAH后CVS发生的机制尚不明确,可能与内皮素-1(endothelin-1,ET-1)产生、炎性反应、血管收缩/舒张功能障碍等有关[3-4]。目前,SAH患者主要通过术中应用溶栓剂、术后应用血管扩张剂、抗炎治疗等预防和治疗CVS,取得了一定的疗效,但仍有部分患者的疗效不理想[5]。利多卡因是一种酰胺类局麻药,临床常用于局部麻醉和抗心律失常[6]。近年来研究发现,利多卡因可以保护神经细胞离子通道,调节脑组织氧自由基能量代谢,发挥脑保护作用,对大鼠SAH后迟发性CVS具有一定的保护作用,但其作用机制尚未明确[7]。因此,本研究观察了利多卡因对大鼠SAH后迟发性CVS的影响,并进一步分析了其脑保护作用的可能机制,旨在为SAH后迟发性CVS的防治提供一定的理论依据,报道如下。
1 材料与方法
1.1 试剂与动物 SPF级SD雄性大鼠54只,体质量180~220 g,由北京北方艾特生物科技有限公司提供[动物许可证号SCXK(京)2020-0005],室温下自由摄食与饮水。利多卡因购于国药集团容生制药有限公司;TUNEL组织细胞凋亡检测试剂盒购自艾美捷科技有限公司;超氧化物歧化酶(superoxide dismutase,SOD)、谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-Px)、丙二醛(malondialdehyde,MDA)、ET-1和一氧化氮(nitric oxide,NO)检测试剂盒,均购自上海雅吉生物科技有限公司;p38丝裂原活化蛋白激酶(p38 mitogen activated protein kinase,p38MAPK)、p-p38MAPK、内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)、p-eNOS抗体和辣根过氧化酶(horseradish peroxidase,HRP)标记羊抗兔IgG均购于美国Santa Cruz公司。
1.2 动物造模及分组 实验于2019年2-5月于华北理工大学附属医院神经外科实验室进行。大鼠预饲养1周后,采用随机数字表法分为假手术组、SAH组、利多卡因组,每组18只。SAH组和利多卡因组大鼠采用枕大池注血法复制SAH模型[8],腹腔注射2.5%戊巴比妥钠麻醉,以枕外隆凸为中点行约1.0 cm切口,逐层分离,初步定位枕骨、环椎及环枕膜。分离暴露左侧股动脉,抽取股动脉血0.3 ml,然后将注射器针头沿枕骨下滑至枕骨大孔,并向前推进至枕大池,将0.3 ml血液缓缓注入枕大池,保持俯卧位头低脚高约30 min,缝合切口,并用碘伏进行消毒。48 h后采用同样的方法抽取0.3 ml右侧股动脉血注入枕大池,若造模后大鼠基底动脉管腔缩小,管壁增厚则为造模成功。其中假手术组大鼠暴露股动脉后,将0.3 ml生理盐水缓慢注入枕大池,采取同样的方法注入2次,然后缝合伤口。第二次枕大池注血后30 min,利多卡因组大鼠向枕大池注入2%的利多卡因0.1 ml,SAH组和假手术组注入等体积生理盐水。
1.3 检测指标与方法 造模后3 d、5 d和7 d,每组各取6只大鼠进行组织取材,常规麻醉,经枕骨大孔抽取脑脊液,暂存于-80℃冰箱,向升主动脉插管灌注4%多聚甲醛PBS溶液,分离基底动脉,置入4%多聚甲醛中固定;取脑皮质及海马组织,一部分置于4%多聚甲醛中固定,一部分暂存于-80℃冰箱待测。
1.3.1 基底动脉和脑组织的病理变化观察:取4%多聚甲醛固定的基底动脉组织,进行常规切片制作和HE染色,光镜下(×100)观察基底动脉组织病理变化,随机选取5个视野,采用Image J图像分析系统测量基底动脉管径周长和管壁厚度。取4%多聚甲醛固定的脑海马组织,进行常规切片制作和焦油紫染色,光镜下(×200)观察海马CA1区神经元形态结构。
1.3.2 TUNEL法检测各组大鼠脑组织凋亡情况:取4%多聚甲醛固定的脑海马组织,进行常规切片制作和TUNEL染色,光学显微镜下(×200)观察神经细胞凋亡情况,随机选取10个视野,采用ImageJ图像分析系统计数阳性细胞数与总细胞数,计算细胞凋亡率。
1.3.3 脑脊液中氧化应激和血管收缩因子水平检测:取暂存于-80℃冰箱的脑脊液,采用相应试剂盒测定脑脊液SOD、MDA、GSH-Px、NO和ET-1水平,严格按试剂盒说明书进行操作。
1.3.4 Western Blot测定脑组织中p38MAPK、eNOS蛋白表达:取大鼠脑皮质及海马组织,匀浆,加入1 ml细胞裂解液提取总蛋白,经BCA法蛋白定量,依次进行电泳、转膜、封闭,加入一抗p38MAPK(1∶1 000)、p-p38MAPK(1∶1 000)、eNOS(1∶1 000)、p-eNOS(1∶1 000),以β-actin(1∶500)为内参,4℃下孵育过夜,加HRP标记的二抗(1∶1 000),37℃下孵育2 h,显影,Image J图像分析系统分析蛋白相对表达量。
2 结 果
2.1 各组大鼠基底动脉和脑组织损伤比较 HE染色结果显示,假手术组大鼠基底动脉管腔呈圆形或椭圆形,均未见明显异常;SAH组大鼠基底动脉管腔不规则,管壁增厚,管腔面积缩小,且随时间延长,病变越明显;利多卡因组大鼠基底动脉管腔较规则,有收缩,但相较于SAH组病变明显减轻,见图1。

图1 各组大鼠不同时点基底动脉病理特征比较(HE染色,×100)
焦油紫染色结果显示,假手术组大鼠海马CA1区神经细胞排列整齐形态,未见明显异常;SAH组大鼠海马CA1区神经细胞出现固缩变性,细胞间隙增加,胞浆呈深紫色,且随时间延长,病变越明显;利多卡因组海马CA1区神经细胞出现少量细胞固缩变性,病变较SAH组显著减轻,见图2。

图2 各组大鼠不同时点海马CA1区病理特征比较(焦油紫染色,×200)
2.2 各组大鼠基底动脉管径和管壁厚度的比较 相应干预后第3 d、5 d和7 d,SAH组大鼠基底动脉内径周长小于假手术组,基底动脉壁厚度大于假手术组(P<0.05);干预后第5、7 d ,利多卡因组大鼠基底动脉内径周长大于SAH组,基底动脉壁厚度小于SAH组(P<0.05),见表1。
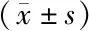
表1 各组大鼠基底动脉管径和管壁厚度比较
2.3 各组大鼠海马组织细胞凋亡的比较 各组大鼠予相应干预后第3 d、5 d和7 d,SAH组大鼠海马组织细胞凋亡率均高于假手术组(P<0.05);利多卡因组大鼠海马组织细胞凋亡率均低于SAH组(P<0.05),见图3、表2。

表2 各组大鼠海马组织细胞凋亡率比较

图3 各组大鼠不同时点海马组织细胞凋亡情况比较(TUNEL染色,×200)
2.4 各组大鼠脑脊液氧化应激和血管收缩因子水平比较 各组大鼠予相应干预后第7d,SAH组大鼠脑脊液SOD、GSH-Px和NO水平低于假手术组,MDA和ET-1水平高于假手术组(P<0.05);利多卡因组大鼠脑脊液SOD、GSH-Px和NO水平高于SAH组,MDA和ET-1水平低于SAH组(P<0.05),见表3。
表3 各组SAH大鼠脑脊液氧化应激和血管收缩因子水平比较
2.5 各组大鼠脑组织p38MAPK及eNOS表达的比较 各组大鼠予相应干预后第7d,SAH组大鼠脑组织p-p38MAPK/p38MAPK蛋白表达高于假手术组(P<0.05),p-eNOS/eNOS蛋白表达低于假手术组(P<0.05);利多卡因组p-p38MAPK/p38MAPK蛋白表达低于SAH组(P<0.05),p-eNOS/eNOS蛋白表达高于SAH组(P<0.05)。见图4。
3 讨 论
SAH后CVS是多种因素作用导致血管平滑肌收缩异常的结果,临床多采用钙离子通道阻滞剂、ET拮抗剂、他汀类药物等治疗,以调节血管收缩和血管炎性反应,达到治疗效果[9]。利多卡因是近年来发现的一种脑保护剂。既往多项研究显示,利多卡因可以阻断神经细胞Na+、K+、Ca2+离子通道,调节神经细胞的氧化应激水平,改善脑组织能量代谢,发挥脑保护作用[10-11]。本研究中,SAH组大鼠脑组织神经细胞病变明显,基底动脉管腔周长明显缩短,动脉壁厚度明显增厚,经利多卡因干预后,神经细胞病变明显减轻,基底动脉管腔周长延长,动脉壁厚度减小,提示SAH大鼠出现明显脑组织损伤和CVS,而利多卡因干预可明显减轻脑组织损伤,缓解CVS的发生。且本研究发现,SAH组大鼠脑组织神经细胞凋亡率明显升高,而利多卡因干预可以明显抑制脑组织细胞凋亡发生。

注:与假手术组相比,aP<0.05;与SAH组相比,bP<0.05
SOD和GSH-Px是机体重要的抗氧化酶,可以清除机体过多的氧自由基,降低过氧化中间产物MDA水平[12]。已有研究显示,SAH大鼠产生的过多氧自由基可以破坏线粒体结构,激活Bax/Bcl-2凋亡途径,引起线粒体能量代谢障碍,导致脑组织损伤[13-15]。本研究中,利多卡因干预可以升高SAH大鼠脑脊液中的SOD和GSH-Px活性,降低MDA水平,提示利多卡因干预可能通过降低SAH大鼠的氧化应激水平,清除脑组织过量的氧自由基,减轻脑组织损伤。NO是一种气态自由基,可以激活细胞内钙泵,减少细胞内游离Ca2+,舒张血管,还可以通过多种途径抑制ET-1的分泌,抑制血管平滑肌细胞收缩,在SAH后CVS的发生中发挥重要的调控作用[16-17]。ET-1是一种主要由血管内皮细胞合成的强效血管收缩因子,可以促进血管平滑肌增殖,还可以抑制eNOS表达,抑制NO合成,促进SAH后CVS的发生[18-19]。Munakata等[20]研究亦显示,NO和ET-1是调控SAH后CVS发生的关键细胞因子。本研究中,利多卡因干预可以升高SAH大鼠脑脊液中的NO水平,降低ET-1水平。提示利多卡因可能通过调节NO和ET-1释放,抑制SAH后CVS的发生。
已有多项研究显示,p38 MAPK信号通路激活可以调节eNOS-NO系统的表达,磷酸化的p38 MAPK可以抑制eNOS的磷酸化,抑制NO的合成和分泌,调节血管收缩功能,参与调控SAH后CVS的发生发展[21-22]。Jiang等[23]的研究显示,利多卡因可以抑制p38 MAPK的磷酸化,减轻脑缺血再灌注大鼠的脑组织损伤。本研究结果表明,利多卡因可能通过抑制p38 MAPK的磷酸化,促进eNOS的表达,调节NO和ET-1释放,缓解SAH后CVS的进展。
综上所述,利多卡因可能通过抑制p38 MAPK的磷酸化,促进eNOS-NO表达,抑制ET-1的分泌,调节血管收缩功能,缓解SAH后CVS的发生发展,还可以抑制脑组织细胞凋亡,减轻脑组织损伤,有望应用于临床SAH后CVS的防治。但本研究尚处于探索阶段,其具体机制尚需进一步实验验证。
利益冲突:所有作者声明无利益冲突
作者贡献声明
苏飞:设计研究方案,实施研究过程,资料搜集整理,论文撰写;陈博文、李向男、刘佳琦:设计研究方案,分析试验数据,论文修改,进行统计学分析;陈扬:提出研究思路,实施研究过程,论文审核
猜你喜欢
中国银幕(2022年4期)2022-04-07 21:28:24
石家庄学院学报(2021年6期)2021-11-26 08:18:06
中成药(2021年5期)2021-07-21 08:39:04
云南医药(2021年3期)2021-07-21 05:40:54
山东工业技术(2016年15期)2016-12-01 05:32:02
中外医疗(2015年5期)2016-01-04 03:58:02
中国当代医药(2015年30期)2015-03-01 02:08:07
发明与创新(2015年37期)2015-02-27 10:40:25
中国卫生标准管理(2015年7期)2015-01-27 05:24:31
癌变·畸变·突变(2014年3期)2014-03-01 04:39:48
