
形貌效应对渣油加氢反应Ni-Mo/Al2O3催化剂预硫化动力学影响
2021-12-22 09:13茆志伟刘铁斌朱慧红
石油学报(石油加工) 2021年6期
刘 哲,茆志伟,冯 翔,刘铁斌,朱慧红,彭 冲
(1.中国石油大学(华东)重质油国家重点实验室,山东 青岛 266580;2.中国石化 大连石油化工研究院,辽宁 大连 116045)
随着原油重质化程度的提高以及环保法的日趋严格,经济有效地处理减压渣油是中国炼油行业进行一体化转型升级的关键之一[1-3]。渣油加氢可以实现渣油资源的高效化和清洁化利用,对渣油加氢工艺以及加氢催化剂的改进已有报道[4-6],其中,沸腾床渣油加氢技术由于具有较强的原料适应性以及较长的运转周期受到了广泛关注[7-8]。在沸腾床渣油加氢工艺中,催化剂的预硫化是至关重要的一步[9-10],直接影响了催化剂活性位结构(金属硫化物),并进而决定了催化加氢的活性。通常而言,预硫化反应在硫化剂和H2存在的条件下进行,催化剂、反应温度和压力、H2S浓度等[9]都会影响预硫化的效果。研究催化剂预硫化过程的动力学并了解预硫化反应的规律和特点,有助于实现催化剂结构的精确调控以及加氢反应性能的有效强化。
加氢催化剂的预硫化属于气-固相非催化反应,现提出的动力学模型主要有均相模型、未反应缩核模型、层式反应模型、粒子模型和孔道模型等[11-15]。目前应用最为广泛的是由Szekely等[12]提出的粒子模型。孙征[16]研究预硫化本征和宏观动力学时分别采用了未反应缩核和等效粒子模型。研究表明,未反应缩核模型适用于硫化反应的本征动力学过程,而等效粒子模型构建的宏观动力学准确性较高。Ho和Reyes[17]研究Co-Mo催化剂的预硫化反应动力学时发现,预硫化反应对H2S为一级反应,对固体为零级。
尽管加氢催化剂预硫化动力学的相关研究已经有所报道,但是对形貌效应影响下催化剂预硫化反应中活性组分的硫化程度缺少相应的研究。中国石化大连石化研究院自主研发了STRONG沸腾床渣油加氢技术,该技术以微球形催化剂替代了传统沸腾床中的圆柱形催化剂,具有原料适应性强、传热和传质性能优以及运转周期长等优点[18-20]。由此可见,加氢催化剂宏观形貌和结构尺寸的不同会直接影响反应性能。因此,探究催化剂的形貌效应对预硫化动力学的影响很有必要。笔者采用粒子模型对Ni-Mo/Al2O3催化剂预硫化反应过程进行颗粒宏观动力学建模,并研究了催化剂的形貌、尺寸和孔隙率等结构因素对金属组分MoO3预硫化反应速率和总体转化率的影响。
1 实验部分
1.1 原料和试剂
碱式碳酸镍(质量分数98%)和三氧化钼(质量分数99.9%),上海易恩化学技术有限公司产品;二甲基二硫醚(质量分数98%),国药集团化学试剂有限公司产品;减压渣油、γ-Al2O3载体、黏结剂和助剂,取自中国石化大连石化研究院。
1.2 催化剂的制备
2种不同宏观形貌(微球形和圆柱形)的渣油加氢Ni-Mo/Al2O3催化剂的详细制备步骤如下:首先,将一定量的NiCO3·2Ni(OH)2·4H2O和MoO32种原料分别溶于一定浓度的磷酸水溶液中,加热沸腾至全部溶解,并将2种溶液共同浸渍到γ-Al2O3载体上。随后,浸渍12 h,并在120 ℃烘箱中干燥8 h,并在800 ℃马弗炉中焙烧4 h,从而得到需要的大孔孔道以及合适的孔体积。圆柱形的Ni-Mo/Al2O3催化剂采用传统的挤压成型工艺制备,而微球形的Ni-Mo/Al2O3催化剂为中国石化大连石化研究院研发的STRONG沸腾床成型工艺制备[21]。在2种形貌催化剂成型的过程中,所使用的γ-Al2O3载体、黏结剂和助剂均保持相同。与圆柱形催化剂相比,制备得到的微球形催化剂尺寸更小,孔隙率更大。
1.3 催化剂的程序升温硫化实验(TPS)
微球形和圆柱形催化剂的预硫化反应测试在程序升温硫化装置上进行。每次实验过程中,首先在石英样品管中装填0.3 g成型的催化剂,随后以5 ℃/min的升温速率将温度从室温升到120 ℃,在He气氛下吹扫0.5 h。随后,将温度以10 ℃/min的升温速率升至320 ℃,通入5%H2S和95%H2(体积分数)的混合气对催化剂进行硫化处理,流速设置为15 mL/min,硫化反应时间持续2 h,采用质谱对H2S进行检测。催化剂上活性组分MoO3的转化率采用式(1)进行计算:
(1)
2 结果与讨论
2.1 预硫化动力学建模
2.1.1 模型的建立
图1为粒子模型示意图。如图1所示,常规粒子模型假设固体反应物为一个多孔的大颗粒,其由大量无孔的微小球形粒子组成,粒子间的空隙组成了颗粒的孔道。气体反应物进入颗粒后与粒子接触,并先与颗粒外边缘处的粒子发生反应。当反应还未完全结束时,气体就会扩散到颗粒内部,接触到内部的粒子并同时发生反应,此时颗粒由外而内每一部分的粒子反应进度都是不同的。在气体与粒子反应的过程中,气体需要扩散通过粒子外部产生的产物层,由于产物层中的气体扩散很慢,使得晶粒与气体间的化学反应过程可以采用未反应缩核模型进行描述。
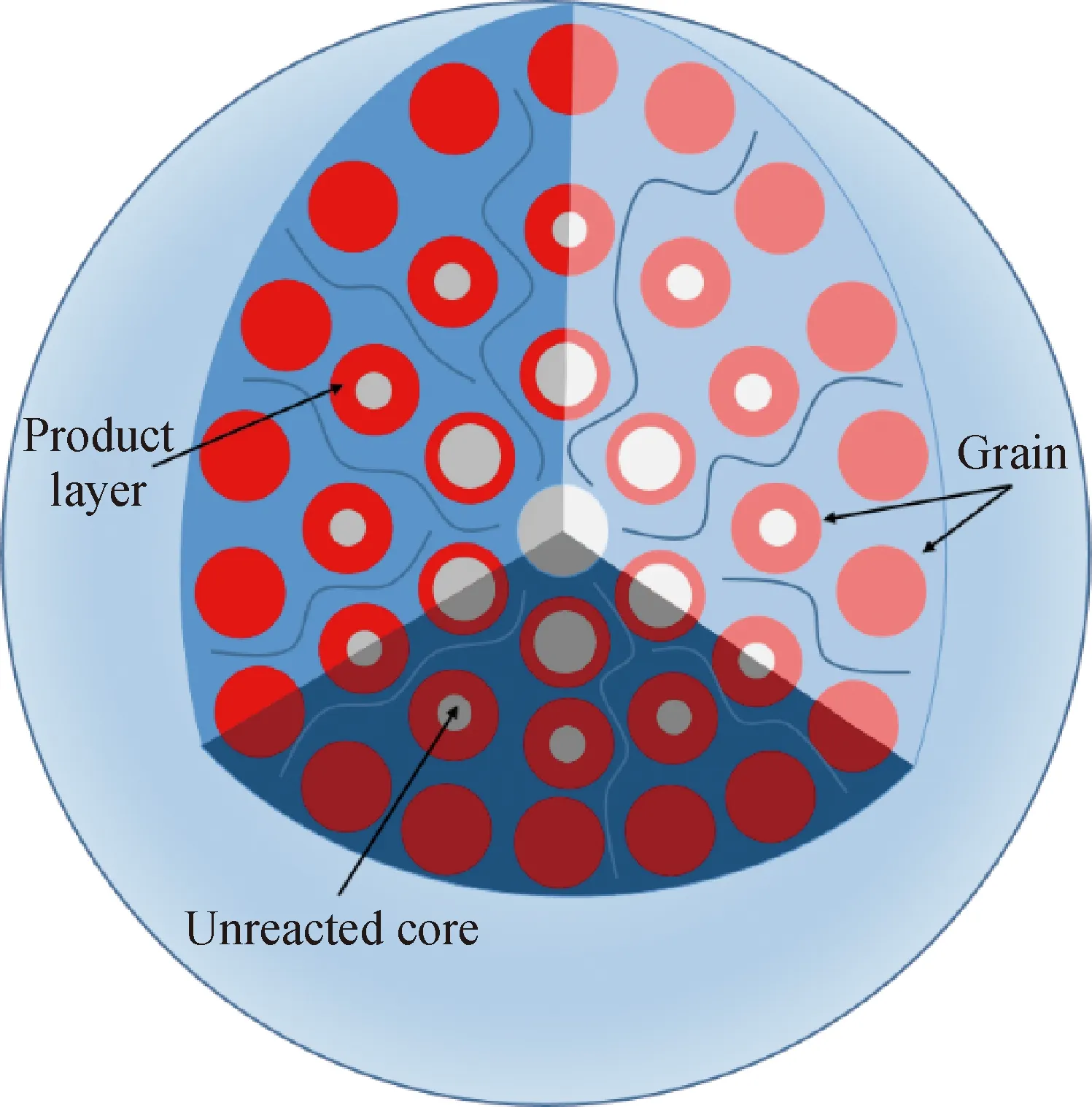
图1 粒子模型示意图Fig.1 Schematic diagram of grain model
由于渣油加氢催化剂预硫化过程中活性金属组分的反应属于气-固相非催化反应,因此γ-Al2O3载体作为一种惰性反应物在硫化过程中不与H2S和H2发生反应。Ni-Mo/Al2O3催化剂中NiO助剂含量较少,活性组分MoO3是主要的固体反应物,可以假设MoO3均匀分布在氧化铝载体上。H2S和H2从孔道扩散进入反应物颗粒内部,与活性金属组分粒子MoO3发生反应,反应方程为:
MoO3(s)+2H2S(g)+H2(g)→
MoS2(s)+2H2O(g)
(2)
2种不同形貌(微球形和圆柱形)催化剂的结构参数统计列于表1。
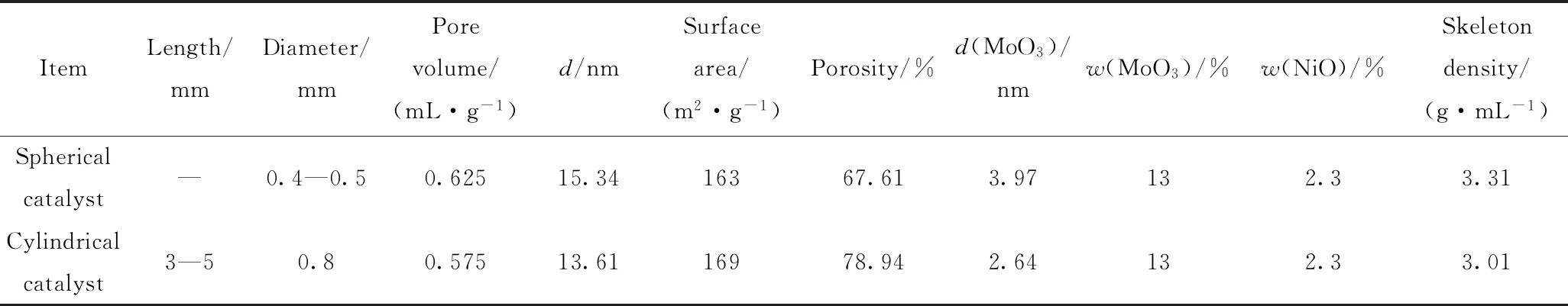
表1 微球形和圆柱形Ni-Mo/Al2O3催化剂的结构参数Table 1 Structural properties of spherical and cylindrical Ni-Mo/Al2O3 catalysts
在粒子模型中作以下假设:
(1)反应过程中γ-Al2O3载体颗粒的大小是恒定不变的,NiO含量忽略不计,将MoO3视为粒子,由外而内均匀分布在γ-Al2O3载体中。
(2)由于反应物MoO3和产物MoS2的摩尔体积相近,因此假设粒子反应前后尺寸不变,即γ-Al2O3载体的孔道结构和孔隙率不随反应发生变化。
(3)γ-Al2O3载体不参与反应,并且在反应过程中,整个催化剂颗粒和单个MoO3粒子内的温度梯度均不予考虑,即颗粒和粒子均处于等温状态。
(4)H2S从气相主体到γ-Al2O3载体外表面的外扩散阻力忽略不计,H2相对于H2S大大过量,因此忽略H2量的变化,总体反应速率主要取决于气体反应物H2S在颗粒孔道内的扩散速率、在粒子产物层中的扩散速率以及H2S与MoO3的表面化学反应速率。
(5)反应对于H2S为一级不可逆反应,固体反应物的反应级数为零级。
(6)在催化剂颗粒内的H2S气体扩散浓度为拟稳态,并且假设H2S在MoS2产物层中的扩散系数是MoO3总体转化率的函数。
(7)MoO3粒子中的硫化反应过程采用未反应缩核模型进行描述。
(8)不同形貌的催化剂具有不同的形状因子,微球形催化剂为3,圆柱形催化剂为2;不同形貌催化剂的当量半径r0采用式(3)计算。
r0=FpVp/Ap
(3)
根据以上假设对Ni-Mo/Al2O3催化剂的预硫化过程进行颗粒宏观动力学建模,如图2所示,以球形催化剂颗粒为基础,拟稳态的假设下,在距离催化剂颗粒中心为r的位置处取一厚度为Δr的微元球壳,以A代表气体反应物H2S,S代表固体反应物MoO3,当气体反应物扩散进入催化剂的孔道时,对微元球壳中的H2S进行物料衡算。定态下,单位时间内扩散进入和离开微元壳体的反应物A的量表示为式(4)和式(5),单位时间内在壳体内化学反应消耗的A的量为式(6)。
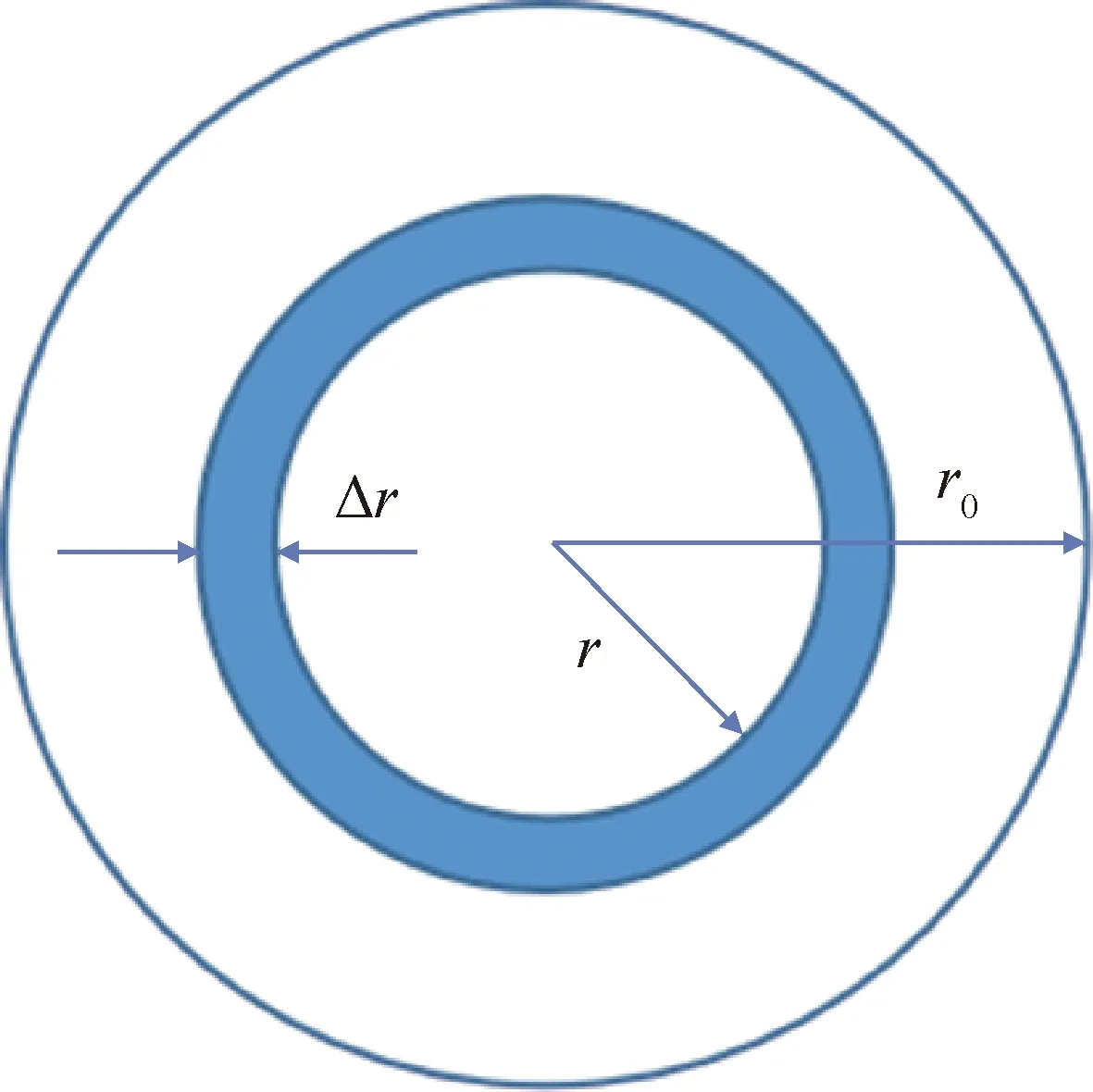
图2 球形催化剂中微元壳体Fig.2 Micro-element shell in spherical catalyst
(4)
(5)
nAc=rA4πr2Δr
(6)
根据微分中值定理,r+Δr处的反应物摩尔浓度可由式(7)计算。
(7)
将式(7)代入扩散进量式(4)中可得:

(8)
扩散进量式(8)减去扩散出量式(5)即为单位时间内在壳体内化学反应消耗的A的量,略去高阶无穷小(Δr)2和(Δr)3并且联立式(6)可得:
(9)
即:
(10)
将催化剂颗粒的形状因子Fp考虑在内,即可得到:
(11)
式(11)为颗粒内反应物H2S的物料衡算表达式,其边界条件为:
(12)
r=r0,CA=CA0
(13)
根据未反应缩核模型,对于单位体积催化剂颗粒,MoO3粒子的占有体积分数为:
(14)
则气体组分A的反应速率rA为:
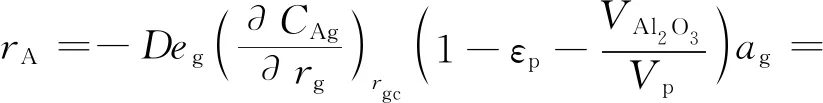
(15)
根据式(2)化学反应方程中反应物的化学计量比,气体反应物A的反应速率为固体反应物S的2倍,即:
(16)
由于:
(17)
(18)
则:
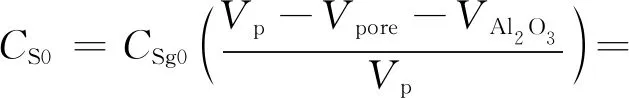
(19)
此外:
CS=CS0(1-xg)
(20)
则式(16)可以转化为:

(21)
为了获得MoO3粒子反应界面的气体浓度CAgc和颗粒内气体浓度CA的关系,建立粒子产物层中偏微分物料衡算式,粒子产物层中H2S只进行扩散而不发生反应,故可得:
(22)
边界条件为:
rg=rg0,CAg=CA
(23)
(24)
且在粒子产物层和未反应内核界面位置rgc处,单位反应界面上的气体扩散速率与反应速率恒等:
(25)
根据边界条件对式(22)进行2次积分可以得到CAg在粒子内的径向浓度分布关系式:
(26)
则粒子在反应界面上的气体反应物浓度CAgc为:
(27)
将粒子反应界面的气体浓度CAgc代入到式(25)中,可得:
(28)
将式(28)结果代入到式(15)中,可得:
(29)
联立式(19)、式(21)和式(29)最终可以得到:
(30)
CA=CAg0
(31)
(32)
(33)
式(30)为催化剂颗粒MoO3的局部转化率xg随时间t和气体浓度CA的变化方程。将由式(11)计算得到的气体CA与r的关系代入到式(30)中,通过MATLAB计算可以得到xg与时间t和距离r的关系,最后通过式(34)得到催化剂颗粒中MoO3总体反应转化率X与时间t的关系:
(34)
在以上建立的预硫化反应粒子模型中,很多参数需要借助辅助公式进行计算,以下为所使用到的相关辅助公式:模型假设(6)中的产物层扩散系数是总体转化率X的函数,可以用式(35)表示:
Deg(X)=Deg0exp(-aX)
(35)
催化剂孔道中H2S的有效扩散系数De采用式(36)计算:
(36)
式(36)中的气体扩散系数De1由分子扩散系数DH2S和努森扩散系数DK组成,如式(37)所示:
(37)
其中,DK表示气体分子与孔道壁的碰撞,计算公式为式(38)~式(40):
(38)
(39)
(40)
DH2S计算公式为式(41):
(41)
该模型中所用本征反应速率常数ks和反应级数n均引用自孙征[16]对于加氢催化剂预硫化动力学的研究结果,结果表明硫化反应为一级不可逆反应,初始反应速率常数表达式为:
干线公路在发挥其原有功能的基础上兼顾了城市道路功能,致使城乡区域没有形成一个连续的空间体系,城乡结合部交通安全问题突出。
(42)
2.1.2 模型的求解
催化剂颗粒中反应物H2S的浓度分布曲线可以由式(11)及其边界条件经过解析法求得,而粒子内转化率xg则需要采用Runge-Kutta数值求解方法对式(30)计算,最后可以用来计算式(34),获得粒子MoO3总体转化率X随着时间t的变化曲线。在计算过程中时间步长选择为0.002 min,这可以满足H2S在颗粒孔道内的拟稳态假设。图3所示为MATLAB软件计算算法的流程示意图。
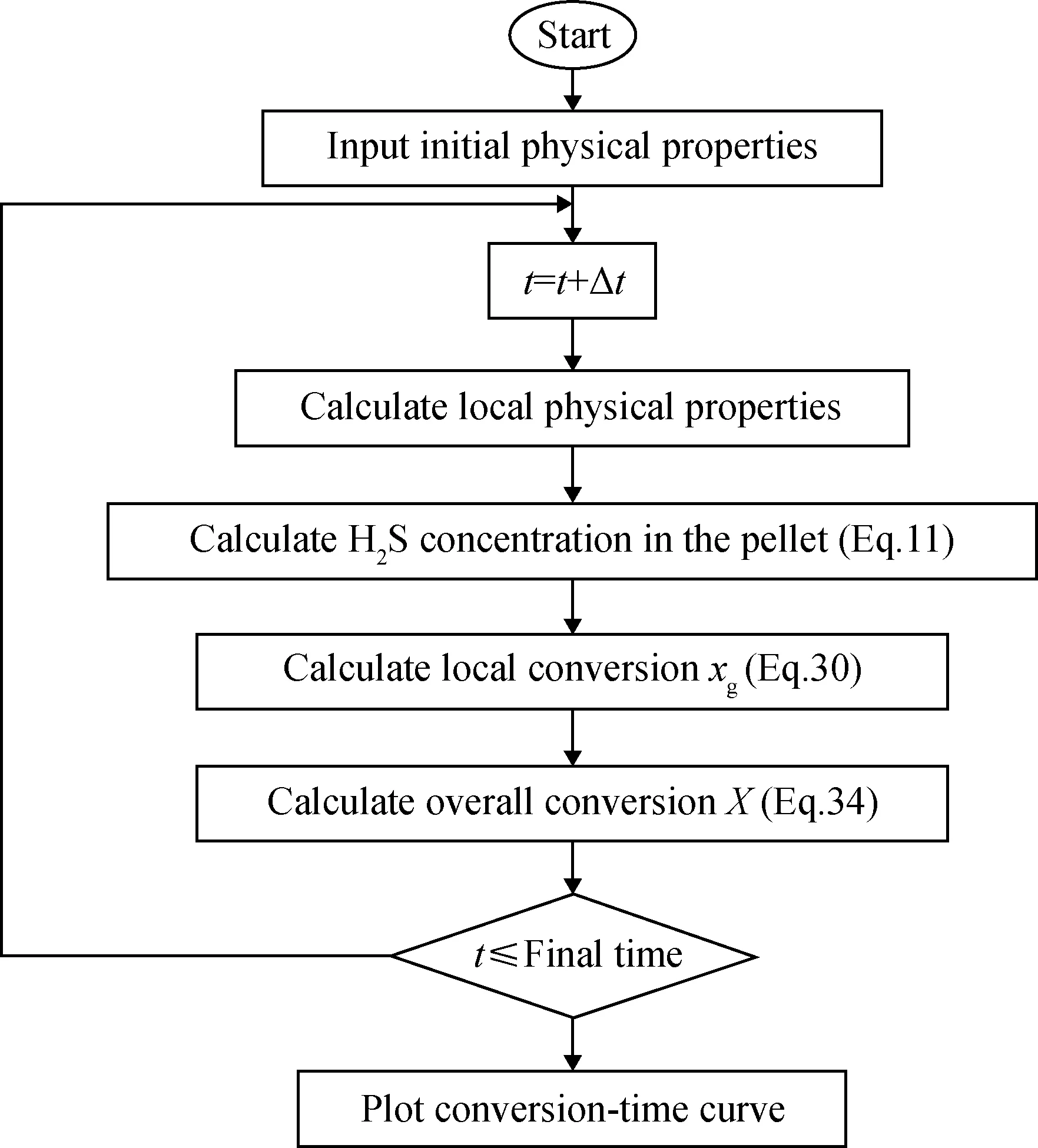
图3 MATLAB算法流程示意图Fig.3 Schematic diagram of MATLAB algorithm
式(11)的求解过程如下:联立式(11)和式(29)2个公式,可以得到:

(43)
当催化剂颗粒为圆柱形时,即形状因子Fp为2,式(43)变为:
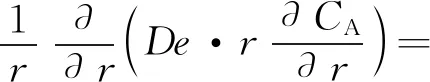
(44)
当催化剂颗粒为球形时,即形状因子Fp为3,式(43)变为:
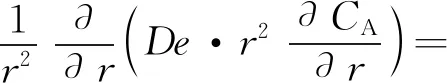
(45)
对于式(35)而言,Deg0是初始产物层扩散系数,a是拟合参数,a的数值是采用Nelder-Mead算法进行优化,回归参数是通过最小化目标函数进行的:
(46)
式中:上标exp为实验值,cal为计算值。
2.1.3 模型的验证
为了验证建立的Ni-Mo/Al2O3加氢催化剂预硫化动力学模型对金属活性组分MoO3硫化反应规律的揭示和预测的准确性,在320 ℃的反应温度下,对2种不同形貌(微球形和圆柱形)的催化剂进行了程序升温硫化实验,实验获得的Ni-Mo/Al2O3催化剂上金属活性组分MoO3的转化率与反应时间的关系如图4中散点所示。随后,引用文献[16]中已报道的预硫化本征动力学反应速率常数ks和反应级数n作为基础来进行动力学规律的研究。采用上述粒子模型对活性金属组分MoO3转化率与时间关系进行了计算,模型计算的结果如图4中实线所示。
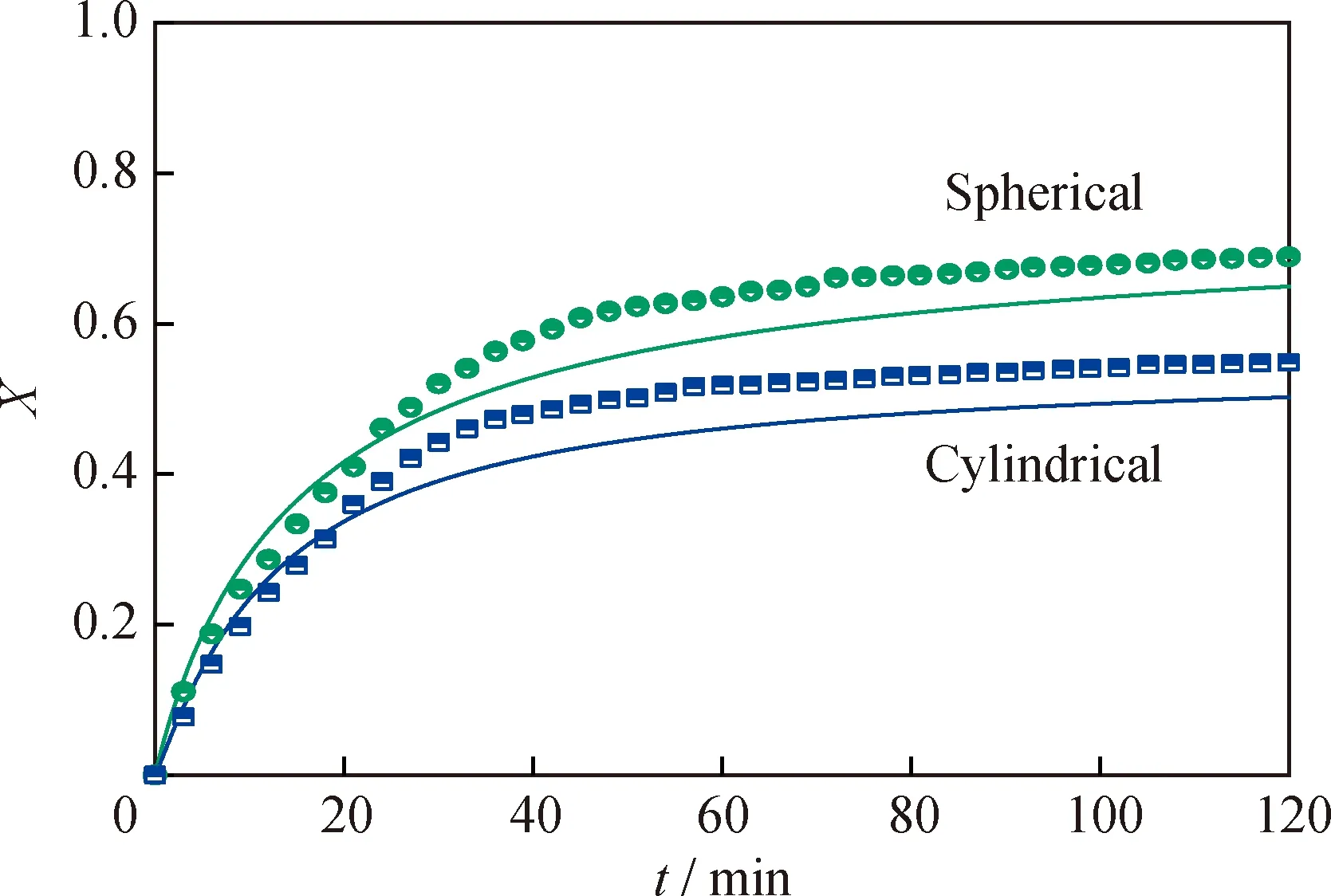
Green:Spherical catalyst;Blue:Cylindrical catalyst;Points:Experimental data;Line:Modeling results图4 不同形貌催化剂预硫化实验数据与模拟数据对比Fig.4 Comparison of experimental and modeling results for pre-sulfurization of catalysts with different morphologiesReaction conditions:m(Catalyst)=0.3 g;V(H2S)∶V(H2)=1∶19;T=320 ℃;p=0.1 MPa;Reaction time:2 h;Flow rate of feed gas:15 mL/min
从图4可以对比看出,微球形和圆柱形催化剂硫化实验所得的转化率曲线与模拟计算所得的转化率曲线在总体趋势上保持一致,活性金属组分MoO3的转化率均随着时间增加先快速增长,随后增长速率逐渐放缓。实验结果与模拟结果存在一定偏差,主要是由于在模拟计算过程中着重考虑了MoO3的转化,而假设低含量助剂NiO的影响较小。相关文献[16]中报道,NiO在硫化反应时的活化能比MoO3更高,计算所使用的反应速率常数ks也有所区别,这使得2种催化剂实验所得的初始反应速率比模拟计算值略低。此外,NiO在硫化反应中的转化同样会消耗H2S,而且镍的存在会对钼物种的硫化起到促进作用,而在建模的过程中忽略了镍的影响,因而在实验计算时会一定程度上高估MoO3的转化率,这使得模拟计算的MoO3最终转化率略低于实验所得结果。模型验证结果表明:尽管实验与模拟结果存在一定偏差,但是MoO3的总体转化率规律保持一致,因此该模型可以用于后续加氢催化剂上活性金属组分MoO3硫化反应规律的定性考察。
2.2 预硫化反应影响因素分析
不同形貌催化剂的加氢性能受催化剂床层膨胀率、渣油分子扩散性能以及活性位的共同影响[22-23],在预硫化过程,催化剂的形貌、尺寸、孔隙率都会影响活性位的生成。笔者采用粒子模型对加氢催化剂预硫化反应过程进行颗粒宏观动力学建模,探讨了催化剂的形貌、尺寸和孔隙率对金属活性组分MoO3预硫化反应速率和总体转化率的影响。
2.2.1 催化剂形貌的影响
从图4可以看出,反应初期40 min内转化率曲线斜率较高,这表明硫化反应速率较高。这主要是由于粒子产物层在反应初期并未形成,暴露的活性金属反应物MoO3可以与气体反应物H2S直接接触并发生反应。随着反应的进行,粒子产物层MoS2逐渐形成,气体反应物在产物层中的扩散受阻,反应物H2S难以接触到粒子内部的MoO3,转化率曲线趋于平衡。由此可以推断,硫化反应初始阶段为化学反应控制,中后期转化为产物层扩散控制。
模拟计算得到的微球形和圆柱形催化剂中MoO3粒子的产物层扩散系数随总体转化率的变化曲线如图5所示。从图5中可以看出,反应初始阶段产物层扩散系数最大,随着反应的进行,产物层扩散系数快速下降。这表明初始反应速率最快,气体在产物层中的扩散性能随着反应的进行显著下降,反应速率也急剧下降,因此硫化反应由初始的反应控制转变为后期的扩散控制。通过对比微球形和圆柱形催化剂的拟合结果可以看出,微球形催化剂中MoO3粒子的初始产物层扩散系数Deg0更大,表明反应物在产物层中的渗透性更好,初始反应速率更高。此外,微球形催化剂的产物层对气体反应物H2S的扩散影响更小,从而提高了金属活性组分MoO3的反应速率和总体转化率。
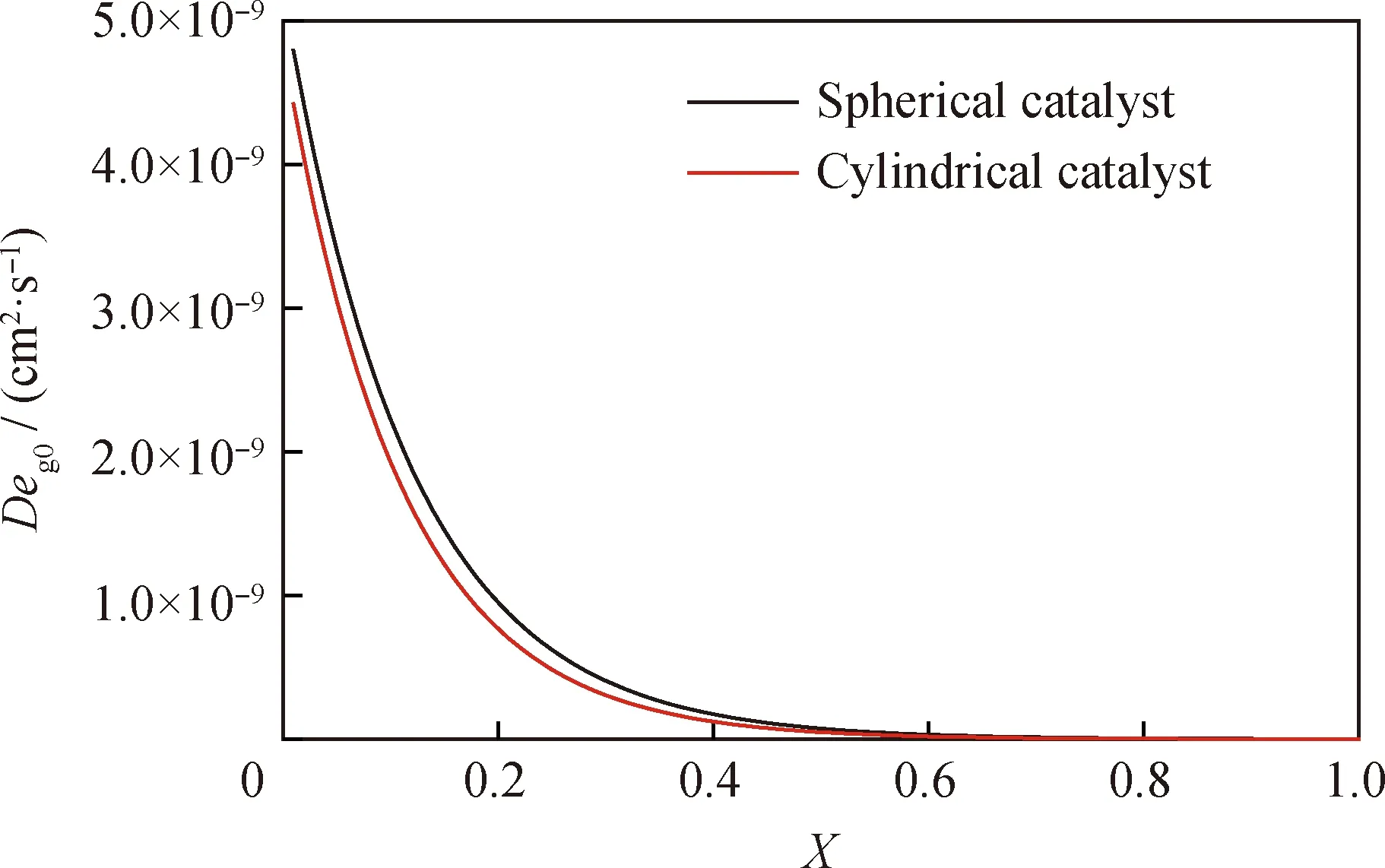
图5 微球形和圆柱形催化剂产物层扩散系数变化曲线Fig.5 Product layer diffusivity curves of spherical and cylindrical catalyst
2种不同形貌催化剂的模拟和实验结果表明:当其他反应条件保持一致时,微球形催化剂具有更高的硫化初始反应速率和总体转化率。与圆柱形催化剂相比,微球形催化剂具有较大的孔隙率和较小的结构尺寸,这可以使反应物H2S更好地扩散进入催化剂的孔道中,与MoO3粒子接触并发生反应。此外,通过H2-TPR、HRTEM、XPS等多种表征手段对2种不同形貌的催化剂进行了表征[22],结果表明,Mo在球形催化剂上与载体间相互作用较弱,易形成容易硫化的六配位Mo6+,使其硫化程度更高。
2.2.2 颗粒尺寸的影响
催化剂的颗粒尺寸对催化剂上活性组分的转化具有重要的影响。宏观的颗粒尺寸大小通常会影响气体反应物在催化剂颗粒中的内扩散,图6所示为球形催化剂宏观颗粒尺寸对活性组分MoO3硫化反应影响规律的粒子模型模拟结果。从图6中可以看出,随着催化剂颗粒尺寸的增大,MoO3硫化反应的初始反应速率和总体转化率均下降,这与Sedghkerdar等[24]在CaO碳酸化反应研究中的模拟结果相一致。

图6 催化剂颗粒尺寸(d)对微球形催化剂上MoO3转化率的影响Fig.6 Effect of catalyst particle size on MoO3 conversion on spherical catalyst
2.2.3 颗粒孔隙率的影响
催化剂颗粒的孔隙率是影响气-固相非催化反应中固体反应物反应速率和转化率的重要参数。催化剂颗粒的孔隙率也影响气体反应物在催化剂孔道中的扩散,通常孔隙率越大,气体在孔道内的扩散系数越大,越容易与固体反应物接触并发生反应。图7所示为球形催化剂颗粒孔隙率对活性组分MoO3硫化反应影响规律的粒子模型模拟结果。从图7中可以看出,MoO3的初始反应速率和总体转化率随着催化剂颗粒孔隙率的增大而增大,孔隙率为0.78的球形催化剂具有最高的化学反应速率和转化率。此外,催化剂的孔隙率越大,其对MoO3初始反应速率和总体转化率的影响程度越小。

图7 催化剂颗粒孔隙率对微球形催化剂上MoO3转化率的影响Fig.7 Effect of catalyst particle porosity on MoO3 conversion on spherical catalyst
综上,具有较大的孔隙率和较小颗粒尺寸的微球形催化剂更有利于金属活性组分MoO3的转化。这不仅为不同形貌加氢催化剂预硫化性能和加氢催化性能的差异[23]提供理论解释,还为工业加氢催化剂的精准设计和开发提供理论依据。
3 结 论
(1)采用粒子模型对渣油加氢Ni-Mo/Al2O3催化剂预硫化反应进行颗粒宏观动力学建模与求解,得到了微球形和圆柱形催化剂上活性组分MoO3的转化率与反应时间的关系。
(2)转化率曲线规律表明,硫化反应初期为化学反应控制阶段,粒子产物层并未形成,MoO3与H2S直接接触反应;硫化反应中后期为产物层扩散控制阶段,粒子产物层MoS2生成,H2S扩散受阻,难以到达反应界面。
(3)MoO3粒子的初始硫化反应速率和总体转化率随着催化剂颗粒尺寸的减小以及催化剂颗粒孔隙率的增大而增大。实验和模拟结果均表明,具有较大孔隙率和较小结构尺寸的球形催化剂具有更高的初始反应速率和总体转化率。
符号说明:
ag——粒子界面表面积与粒子体积之比,cm-1;
A——产物层拟合参数,无量纲;
Ap——颗粒外表面积,cm2;
CA——气体H2S在催化剂颗粒孔道内的摩尔浓度,mol/cm3;
CA0——气体H2S在体相中的摩尔浓度,mol/cm3;
CAg——气体H2S在粒子中的摩尔浓度,mol/cm3;
CAgc——气体H2S在粒子中反应界面处的摩尔浓度,mol/cm3;
CAg0——气体H2S在粒子外的摩尔浓度,mol/cm3;
CS——催化剂颗粒中MoO3摩尔浓度,mol/cm3;
CS0——催化剂颗粒中MoO3初始摩尔浓度,mol/cm3;
CSg——MoO3粒子的摩尔浓度,mol/cm3;
CSg0——MoO3粒子的初始摩尔浓度,mol/cm3;
De——气体H2S在颗粒内的有效扩散系数,cm2/s;
De1——气体扩散系数,cm2/s;
DK——努森扩散系数,cm2/s;
DH2S——分子扩散系数,cm2/s;
Deg——产物层扩散系数,cm2/s;
Deg0——初始产物层扩散系数,cm2/s;
Fp——形状因子;
k——进料气组分数目;
ks——粒子单位面积反应界面上的初始反应速率常数,cm4/(mol·s);
K0——比例系数;
mt——颗粒的总质量,g;
MS——MoO3摩尔质量,g/mol;
MW——分子的摩尔质量,g/mol;
nAi——单位时间扩散进入微元球壳的反应物A物质的量,mol;
nAo——单位时间扩散离开微元球壳的反应物A物质的量,mol;
nAc——单位时间在微元球壳内化学反应消耗的反应物A物质的量,mol;
nd——颗粒中粒子的数目,cm-3;
nS——MoO3的物质的量,mol;
nH2S——H2S的消耗量,mol;
nMoO3——催化剂上MoO3的负载量,mol;
NP——实验数据点数目;
OF——最小化目标函数;
p——压强,Pa;
r——颗粒的径向坐标,以颗粒中心为r=0计,cm;
r0——固体催化剂颗粒的当量半径;
rA——单位颗粒体积的粒子上H2S的摩尔消耗速率,mol/(cm3·s);
rg——粒子的径向坐标,以粒子中心为rg=0计,cm;
rg0——粒子的初始半径,cm;
rgc——粒子中未反应内核和产物层界面处的位置,cm;
rS——单位颗粒体积的粒子上MoO3的摩尔消耗速率,mol/(cm3·s);
Rg——气体常数,J/(mol·K);
t——时间,s;
T——温度,K;
VAl2O3——氧化铝载体的体积,cm3;
VPore——孔道体积,cm3;
Vm,s——MoO3的摩尔体积,cm3/mol;
Vp——颗粒总体积,cm3;
φMoO3——单位体积催化剂中MoO3体积分数;
wS——MoO3质量分数,%;
xg——颗粒内活性相MoO3的局部转化率;
X——颗粒内活性相MoO3的总转化率;
εp——颗粒孔隙率;
Δr——球形催化剂颗粒模型中微元球壳的厚度,nm;
δg——拟合参数,无量纲。
猜你喜欢
弹性体(2022年3期)2022-11-15
机械工业标准化与质量(2022年6期)2022-08-12
矿产综合利用(2020年1期)2020-07-24
中国特种设备安全(2019年3期)2019-04-22
中学化学(2017年5期)2017-07-07
中国调味品(2017年2期)2017-03-20
山东工业技术(2016年15期)2016-12-01
中学化学(2016年4期)2016-05-30
中学化学(2015年2期)2015-06-05
理科考试研究·高中(2014年8期)2014-10-17
