
原位干胶合成Ni/SAPO-11催化剂中硅含量及配位环境的调控
2021-12-22 06:55吕玉超于洙末战威龙李富让刘欣梅
石油学报(石油加工) 2021年6期
吕玉超,于洙末,战威龙,李富让,范 磊,杨 野,刘欣梅
(中国石油大学(华东)重质油国家重点实验室,山东 青岛 266580)
烷烃临氢异构化是将正构烷烃转化为异构烷烃的工艺过程[1-2],其中轻质烷烃异构化是生产高辛烷值汽油的重要途径,且异构化油具有不含烯烃、芳烃及硫化物的优点,满足清洁燃料的质量标准。因此,提高汽油池中异构化油含量对推动中国汽油质量升级意义重大。通过烷烃临氢异构化生产异构化油的关键在于高效催化剂的开发,目前工业上广泛采用的是含有贵金属的双功能催化剂,按载体不同可分为氯化铝型、分子筛型及固体超强酸型催化剂,按不同使用温度可分为中温型(分子筛型)和低温型(氯化铝、固体超强酸)催化剂[3]。其中,以氯化铝为载体的低温型催化剂活性强、选择性高,但该催化剂对原料中的水和氧含量十分敏感,原料适应性较差;而以固体超强酸为载体的催化剂,虽对原料中的水和氧含量要求较低,但其热稳定性较差,酸中心易流失。相比之下,分子筛型催化剂由于具有原料杂质耐受程度高、能完全再生等优点而被广泛研究[4]。该类催化剂所用金属组分主要包括Pt、Pd及Ni等[5-8],分子筛主要有Y型分子筛[9]、β分子筛[10]、ZSM-5/22分子筛[11]、丝光沸石[12]和SAPO-n分子筛[13-14]等。目前广泛应用的贵金属异构化催化剂的活性和选择性虽然较高,但其价格昂贵,且易中毒失活,而非贵金属催化剂存在活性低和选择性差的严重缺陷。因此,制备高效和低成本的异构化催化剂是发展烷烃临氢异构化技术的关键。
由烷烃在催化剂表面的双功能反应机理可知[15],催化剂的活性和产物选择性不仅取决于金属与酸性载体的性质,还与金属位和酸性位的协同作用密切相关。中强Brönsted酸是催化异构化反应的活性中心,强酸易加剧裂化反应的发生,降低异构烃选择性。其中,SAPO-11分子筛由于具有独特的一维十元环孔道结构及灵活可调的酸性质,而常被用做烷烃临氢异构化催化剂的载体[16]。笔者前期通过原位干胶法合成了Ni/SAPO-11催化剂[17],该方法实现了Ni/SAPO-11催化剂表面金属的高度分散和酸性质调控,其在正己烷临氢异构化反应中表现出可与Pt/SAPO-11催化剂相媲美的催化性能。这主要是因为该催化剂表面具有较高的活性位数量,过量的金属位使得烯烃中间体及时加氢,减少了裂化反应的发生,进而提高了产物选择性。该催化剂中的过量金属位使发生在酸性位的骨架异构过程成为整个反应的限速步骤,此时调控分子筛骨架Si含量及配位环境是增强载体酸性和提高催化剂性能的关键。
SAPO分子筛的酸性位源于Si原子对磷铝分子筛骨架P或Al原子的同晶取代。Si原子嵌入分子筛骨架的取代机理有2种,分别为SMⅡ取代和SMⅢ取代。SMⅡ取代是指1个骨架P原子被1个 Si原子取代,产生Si(4Al)区域,该取代方式主要生成弱Brönsted酸位;SMⅢ取代是指2个Si原子同时分别取代相邻的P和Al原子,产生硅岛结构(Si(4Si))。当合成凝胶中Si含量较低时,主要发生SMⅡ取代;随着Si含量的增加,SMⅢ取代增多,产生大量硅岛结构,硅岛不产生酸性位,但其边缘的Si(nAl)(n=0、1、2、3)可产生中/强Brönsted酸位[18-19]。因此,调控SAPO分子筛骨架中Si的含量及配位环境是调节SAPO分子筛酸性质的根本途径,提高分子筛骨架硅含量并使其均匀分布是提高分子筛酸量的关键。Yang等[20]考察硅源种类对SAPO-11分子筛骨架硅含量及其分布的影响,结果发现:采用白炭黑制备的SAPO-11分子筛骨架Si含量虽然较低,但其利于小尺寸硅岛结构的生成,分子筛的中强酸位较多;TEOS可提高分子筛骨架中Si含量,但其促进了大尺寸硅岛结构的产生,分子筛酸密度较低;在非水体系中合成SAPO-11分子筛时,TEOS比聚合硅(煅制氧化硅、硅溶胶)更易嵌入SAPO-11分子筛骨架,可以提高骨架Si含量且使硅配位环境呈现多样化,分子筛酸密度较高,负载铂金属后的催化剂Pt/SAPO-11表现出较高的正癸烷异构化活性[21]。Roldn等[22]在合成SAPO-5分子筛时发现,较高的pH值(pH=9.0)有利于Si在骨架中均匀分散,硅岛尺寸较小,分子筛中强酸酸量较高。提高初始凝胶中Si源含量可促进Si原子同晶取代进入分子筛骨架,促进硅岛结构的生成,不利于分子筛酸量的增加。笔者通过控制水溶胶温度实现对SAPO-11骨架Si配位环境的调控,并提出硅物种的迁移、转化机理[23]。升高水溶胶温度可加速多聚态硅物种解聚,提高初始凝胶中活性硅物种浓度,促进硅原子SMⅢ取代,提高硅岛结构含量,Brönsted酸量较低;低水溶胶温度则有利于硅以SMⅡ机理嵌入SAPO-11分子筛骨架,增加Si(4Al,0Si)结构含量,Brönsted酸量增加。表面活性剂也可通过影响硅对骨架磷、铝原子的取代机理,进而改变SAPO分子筛骨架硅的含量与配位环境[24]。Blasco等[25]发现,在己醇和水两相合成体系中引入十六烷基胺可在不改变骨架Si含量的条件下,显著减小SAPO-11分子筛中硅岛结构尺寸,增加Si(nAl,(4-n)Si)(n=0、1、2、3)结构的含量,从而提高分子筛酸量。Guo等[26]发现,与常规晶化法相比,两段晶化法可有效减少SAPO-11分子筛硅岛结构的生成,Si(nAl,(4-n)Si)(n=0、1、2、3)结构含量明显增加。Cui等[27]发现,提高合成体系中硅物种含量在增加骨架含量的同时,也将促进硅岛结构的产生,存在最佳硅物种含量使得SAPO-11分子筛酸量达到最大值。
已有研究大多集中于对分子筛中Si含量及配位环境的调控,直接对催化剂的研究较少。分子筛的合成常采用水热合成法或干胶合成法。水热合成法是指以水作为分子筛晶化的介质,按一定比例混合反应原料制备溶胶,转移溶胶至反应釜进行晶化处理的方法。与传统水热合成法相比,干胶法具有晶化时间短、模板剂用量少且产率高等优点,采用干胶制备的SAPO-11分子筛具有骨架Si分布更为均匀、Si岛尺寸较小和分子筛酸量较高等特点[28]。笔者采用原位干胶法制备Ni/SAPO-11催化剂,通过改变初始凝胶中硅源添加量,控制分子筛骨架硅含量及配位环境调节催化剂的酸性质,考察硅添加含量对催化剂表面镍物种的配位环境、尺寸分布及还原性质等的影响,并以正己烷为模型化合物考察催化剂的异构化性能,确立最佳硅添加量与催化剂的构效关系。
1 实验部分
1.1 原料和试剂
磷酸(H3PO4,质量分数85%)、六水合硝酸镍(Ni(NO3)2·6H2O,化学纯)及二正丙胺(n-C6H15N,DPA,化学纯),均为国药集团化学试剂有限公司产品;酸性硅溶胶(固含量为30%(质量分数))为青岛海洋化工有限公司产品;拟薄水铝石(质量分数70%)为烟台恒辉化工有限公司产品;正己烷(n-C6H14,化学纯)为富宇试剂有限公司产品。
1.2 催化剂的制备
在Ni/SAPO-11催化剂的合成中,分别以拟薄水铝石、磷酸、硅溶胶和六水合硝酸镍作为铝源、磷源、硅源和镍源,按n(Al2O3)∶n(P2O5)∶n(DPA)∶n(SiO2)∶n(NiO)∶n(H2O)(以氧化物计)为1.0∶1.0∶1.1∶x∶0.125∶50(x=0.2、0.4、0.6、0.8)投料。取6.6 g拟薄水铝石加入装有40.5 g去离子水的烧杯,将烧杯置于20 ℃水浴中搅拌10 min。向上述混合体系滴加10.4 g磷酸并搅拌2 h后加入5.1 g二正丙胺,继续搅拌2 h使原料混合均匀。继续向烧杯中加入1.8 g(x=0.2,当x取0.4、1.6、0.8时的硅溶胶质量分别为3.6、5.4、7.2 g)酸性硅溶胶并搅拌2 h。最后加入1.6 g六水合硝酸镍,继续搅拌0.5 h。滴加少许二正丙胺,调节混合体系pH值至5.0。将所得溶胶转移至三口烧瓶中,在80 ℃水浴中蒸干水分后,于110 ℃烘箱内烘12 h形成干胶。研磨干胶至粉末状并置于100 mL晶化釜的内置称量瓶中,晶化釜底加入一定量去离子水(0.3 g/g干胶)。晶化釜于200 ℃条件下恒温处理24 h,晶化结束后的白色粉末经去离子水洗涤至中性,100 ℃烘箱内干燥12 h、马弗炉内500 ℃焙烧6 h及氢气还原后(还原温度550 ℃,时间为2 h)即可得到Ni/SAPO-11催化剂。催化剂命名为Ni/SAPO-11-x,除X射线光电子能谱表征和29Si核磁测试为还原前样品外,其他测试样品均为氢气还原后的Ni/SAPO-11催化剂。
作为对比样品,采用等体积浸渍法制备Pt/SAPO-11 催化剂。首先将SAPO-11原粉在300 ℃ 烘箱干燥3 h,然后浸渍于一定浓度的氯铂酸水溶液中,搅拌5 min后于室温静止22 h。将浸渍后的粉末于100 ℃烘箱中干燥2 h,400 ℃马弗炉焙烧4 h即可得到催化剂样品,记为Pt/SAPO-11,铂负载量(质量分数)为0.3%。
1.3 催化剂表征
采用荷兰帕纳科公司的X’Pert PRO X型X射线衍射仪(XRD)测定样品的晶体结构,仪器采用CuKα辐射(λ=0.15418 nm),管电压为40 kV,管电流为40 mA,扫描速率为10 °/min。采用康塔公司的Autosorb-iQ3型自动物理吸附仪对样品进行孔道结构分析。采用Micromeritics ASAP 2920化学吸附仪进行氨气程序升温脱附(NH3-TPD)实验,分析样品的酸量及酸强度分布。采用瑞士布鲁克Advance 400型核磁共振仪对催化剂骨架Si原子的配位环境进行表征。采用Perkin-Elmer公司的Optima 8000等离子体原子发射光谱仪(ICP)分析样品中Si元素含量。采用日本日立电子公司S-4800冷场发射扫描电子显微镜(SEM)对样品的表观形貌进行分析,SEM加速电压为0.5~30 kV。采用JEM-2100型透射电子显微镜(TEM)对样品表面金属物种的形貌与粒径分布进行分析。采用PHI 500型X射线光电子能谱仪(XPS)分析催化剂表面Ni物种的配位环境,采用C 1s 标准能谱(284.8 eV)对结合能谱进行校准。
1.4 催化剂性能评价
以正己烷为模型化合物对催化剂进行异构化性能评价。反应在不锈钢固定床反应器内进行。将1.2 g催化剂装入反应管内,反应开始前在氢气气氛下对催化剂进行还原处理,还原温度为550 ℃,时间为2 h,氢气体积流量为21 mL/min。还原结束待催化剂床层温度降至反应温度(280、300、320、340、360 ℃)后,将正己烷泵入反应器,在压力为2.0 MPa、氢/烃摩尔比为4.0、质量空速为1.0 h-1的条件下进行反应。反应产物采用Agilent 7820A GC进行在线分析,该色谱配有PC-PONA毛细管柱和氢离子火焰监测器(FID)。对色谱分析结果采用面积归一化法计算反应产物分布,正己烷转化率、异己烷选择性及收率的计算如式(1)~式(3)所示。

(1)

(2)

(3)
2 结果与讨论
2.1 催化剂的物相结构和元素组成
由原位干胶法合成的Ni/SAPO-11-x催化剂的XRD谱图如图1所示。由图1可以看出,4种Ni/SAPO-11-x催化剂在2θ为8.1°、9.7°、16.1°、19.9°、21.8°、23.6°处出现了SAPO-11分子筛的特征衍射峰,说明原位干胶法可制得结晶度良好的Ni/SAPO-11催化剂。催化剂的结晶度主要取决于分子筛载体,将Ni/SAPO-11-0.2催化剂的结晶度设定为100%,并以此为基准计算其他催化剂的相对结晶度,结果如表1所示。由表1可以看出,随着初始凝胶中Si质量分数增加,Ni/SAPO-11催化剂的相对结晶度降低,这可能是由骨架硅含量的增加导致的。随着初始凝胶中SiO2/Al2O3摩尔比由0.2上升至0.8,催化剂中Si质量分数由4.2%上升至8.6%,Si/Al摩尔比由0.23上升至0.50(表1)。Si原子取代骨架P原子会打破原有电荷平衡,使骨架带有负电荷;同时,Si原子半径不同于骨架P和Al原子(rAl>rSi>rP),Si原子的同晶取代会使分子筛骨架发生扭曲,降低分子筛结晶度。

(1)Ni/SAPO-11-0.8;(2)Ni/SAPO-11-0.6;(3)Ni/SAPO-11-0.4;(4)Ni/SAPO-11-0.2图1 Ni/SAPO-11-x催化剂的XRD谱图Fig.1 XRD patterns of Ni/SAPO-11-x catalysts

表1 Ni/SAPO-x催化剂的硅与铝含量、硅/铝摩尔比及相对结晶度Table 1 Si and Al content,Si/Al mole ratio and relative crystallinity of Ni/SAPO-x catalysts
2.2 催化剂的孔结构性质分析
Ni/SAPO-11-x催化剂的氮气吸附-脱附等温线和孔结构数据分别如图2和表2所示。由图2可看出:4种Ni/SAPO-11-x催化剂均具有Ⅰ和Ⅳ混合型吸附等温线。样品在p/p0<0.05低相对压力区表现出对氮气的快速吸附,表明Ni/SAPO-11-x催化剂具有典型的微孔结构;而在p/p0>0.4高相对压力区时,Ni/SAPO-11-x催化剂出现了H3型滞后回环,说明Ni/SAPO-11-x催化剂中存在由晶体颗粒堆积产生的介孔,表2中的Ni/SAPO-11-x催化剂孔结构数据也证明这一结论。此外,由表2可看出:随初始凝胶中Si物种质量分数的增加,Ni/SAPO-11催化剂的比表面积并无明显变化,但微孔比表面积及孔体积呈现减小趋势,这可能是由分子筛相对结晶度下降导致的;而催化剂总孔体积增大,这主要归因于介孔数量的增加。

表2 Ni/SAPO-11-x催化剂的孔结构性质Table 2 Pore structure features of Ni/SAPO-11-x catalysts

图2 Ni/SAPO-11-x催化剂的N2吸附-脱附等温线Fig.2 N2 adsorption-desorption isotherms for the Ni/SAPO-11-x catalysts
2.3 催化剂的形貌分析
Ni/SAPO-11-x催化剂的SEM照片如图3所示。由图3可知,原位干胶法合成的Ni/SAPO-11-x催化剂形状不是十分规则,为类球形颗粒,颗粒尺寸分布在5~10 μm范围。与水热合成法不同[28-29],采用原位干胶法合成Ni/SAPO-11-x催化剂时,分子筛前驱体晶化过程中并未与液态水直接接触,水以蒸汽的形式进入凝胶内部,这使得晶化釜内反应物的释放和传质受到限制,生成的晶体颗粒呈现不规则形状且颗粒尺寸分布不均[27]。由图3还可看出,初始凝胶中Si质量分数的改变对Ni/SAPO-11的形貌及粒径分布并未产生明显影响。

图3 Ni/SAPO-11-x催化剂的SEM照片Fig.3 SEM images of Ni/SAPO-11-x catalysts(a1),(a2)x=0.2;(b1),(b2)x=0.4;(c1),(c2)x=0.6;(d1),(d2)x=0.8
2.4 催化剂表面Ni物种配位环境
笔者前期的工作[17]表明,采用原位干胶法制备的Ni/SAPO-11催化剂(对应此文中Ni/SAPO-11-0.4)中存在3种Ni物种:NiO、镍铝尖晶石及骨架镍。不同Si质量分数所对应的Ni/SAPO-11-x催化剂的XPS谱图和对Ni/SAPO-11-0.2 XPS能谱进行高斯拟合谱图如图4所示。由图4可知:4种Ni/SAPO-11-x催化剂分别在856.3(Ni 2p3/2)和 874.4(Ni 2p1/2)eV的信号峰归属于NiO中八面体配位的Ni2+;分别在结合能为857.6 eV(Ni 2p3/2)和875.4 eV(Ni 2p1/2)处的信号峰归属为镍铝尖晶石中Ni2+;分别在结合能为858.7 eV(Ni 2p3/2)和876.5 eV(Ni 2p1/2)处的信号峰表明存在四配位骨架镍。Ni/SAPO-11-0.4催化剂的XPS谱图与其他3个样品接近,表明改变初始凝胶中Si质量分数并不影响Ni/SAPO-11-x催化剂表面Ni物种配位环境,即不同质量分数Si合成得到的Ni/SAPO-11-x催化剂均存在NiO、镍铝尖晶石及骨架镍3种镍物种。

(1)Ni/SAPO-11-0.8;(2)Ni/SAPO-11-0.6;(3)Ni/SAPO-11-0.4;(4)Ni/SAPO-11-0.2图4 Ni/SAPO-11-x催化剂的XPS谱图Fig.4 XPS profiles of Ni/SAPO-11-x catalysts(a)XPS profiles of Ni/SAPO-11-x catalysts;(b)XPS profiles fitting results of Ni/SAPO-11-0.2 catalyst
2.5 催化剂还原性质分析
原位干胶法合成Ni/SAPO-11-x催化剂的氢气程序升温还原曲线(H2-TPR)如图5所示。由图5可知,3种催化剂的H2-TPR曲线均出现3个H2消耗峰,表明镍物种的还原均可分为3个阶段,分别对应3种不同镍物种,这与XPS分析结果相一致。各氢气消耗峰的归属如下:545 ℃的信号归属为NiO的还原;711 ℃处信号归属为镍铝尖晶石的还原;795 ℃处信号归属为骨架镍物种的还原。由各催化剂的H2-TPR曲线还可发现,初始凝胶中Si质量分数的改变对催化剂还原性质并未产生显著影响,且在异构化反应的预还原过程中,仅表面的NiO颗粒可被还原为Ni0,而镍铝尖晶石和骨架镍保持不变。

图5 Ni/SAPO-11-x催化剂的H2-TPR曲线Fig.5 H2-TPR profiles of Ni/SAPO-11-x catalysts
2.6 催化剂表面Ni颗粒尺寸分布
在催化剂表面的镍物种中,仅还原后得到的Ni0是催化烷烃脱氢/加氢的活性中心。因此,为考察初始凝胶中Si质量分数对催化剂表面Ni0颗粒形貌及尺寸分布的影响,采用TEM对Ni/SAPO-11-x催化剂进行表征,结果如图6所示。由图6可以看出,4种催化剂表面的Ni颗粒呈无规则形状,颗粒粒径均分布在10~20 nm范围内,这也说明初始凝胶中Si质量分数的改变对催化剂表面Ni的分散度无显著影响。

图6 Ni/SAPO-1-x催化剂的TEM照片Fig.6 TEM images of Ni/SAPO-11-x catalysts(a)Ni/SAPO-1-0.2;(b)Ni/SAPO-1-0.4;(c)Ni/SAPO-1-0.6;(d)Ni/SAPO-1-0.8
2.7 催化剂的酸性质和硅配位结构
利用NH3-TPD对Ni/SAPO-11-x催化剂的酸性质进行表征,并以Ni/SAPO-11-0.8催化剂为例,借助高斯方程对NH3-TPD曲线进行分峰解迭,得到3个氨气脱附信号,记为PeakⅠ、PeakⅡ和PeakⅢ,分别归属为氨气由SAPO-11表面弱酸、中强酸及强酸位的脱附,结果如图7所示。根据各谱峰面积计算相应酸中心的酸量,结果如表3所示。由图7和表3可知:随着初始凝胶中Si质量分数的增加,催化剂总酸量呈现先增加后减小的趋势,由大到小依次为:Ni/SAPO-11-0.6、Ni/SAPO-11-0.4、Ni/SAPO-11-0.8(≈Ni/SAPO-11-0.2);中、强酸酸量也有类似规律;而弱酸酸量随Si质量分数的增大呈现降低趋势。SAPO分子筛的酸性与骨架Si质量分数及配位环境密切相关,ICP测试结果表明,SAPO-11分子筛骨架Si质量分数随初始凝胶中Si质量分数增加而增大,因此要对Ni/SAPO-11-x催化剂酸性质的变化进行分析,需进一步表征分子筛中骨架Si的配位环境。
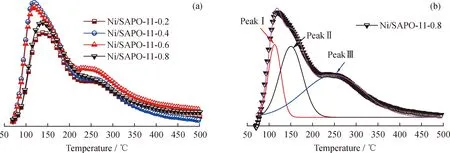
图7 Ni/SAPO-11-x及Ni/SAPO-11-0.8催化剂的氨气程序升温脱附(NH3-TPD)曲线Fig.7 NH3-TPD profiles of Ni/SAPO-11-x and Ni/SAPO-11-0.8 catalysts(a)Ni/SAPO-11-x;(b)Ni/SAPO-11-0.8

表3 Ni/SAPO-11-x催化剂的酸量及酸强度分布Table 3 Acid amounts and distributions for the Ni/SAPO-11-x catalysts
Ni/SPAO-11-x催化剂的29Si MAS NMR谱图如图8所示。借助高斯方程将特征峰解迭为PeakⅠ~PeakⅥ6个信号峰,归属为具有不同配位环境的硅物种[25,30]:PeakⅡ:SAPO区域Si(4Al,0Si),PeakⅠ:SA区域Si(4Al,0Si),PeakⅢ:SA区域Si(3Al,1Si),PeakⅣ:SA区域Si(2Al,2Si),PeakⅤ:SA区域Si(1Al,3Si),PeakⅥ:SA区域Si(0Al,4Si)。对不同配位环境的Si物种对应谱峰面积进行归一化处理,由各Si物种谱峰面积占总谱峰面积的百分含量计算各配位环境Si物种的相对含量,结果如表4所示。由表4可以看出,随着Si含量的增加,Si(4Al,0Si)结构的相对含量整体呈现下降趋势,这是因为合成体系中硅物种含量较低时,Si主要通过SMⅡ取代进入分子筛骨架形成Si(4Al,0Si)结构,提高Si含量则会使Si的取代方式由SMⅡ向SMⅢ转化,Si(nAl,(4-n)Si)(n=0、1、2、3,下同)结构相对含量增大。硅物种含量增加时,硅岛结构的相对含量明显上升,其边界处的Si(nAl,(4-n)Si)结构含量相应增加,过高的Si含量会使硅岛尺寸增大,导致其边界处的Si(nAl,(4-n)Si)含量降低。Ni/SAPO-11-0.2催化剂具有最高质量比例的Si(nAl,(4-n)Si)结构,但其骨架硅含量较低,总酸量较低。随着硅岛相对含量的增加,催化剂的总酸量和中强酸量呈现先增加后降低的趋势,Ni/SAPO-11-0.6的总酸量最高,这与NH3-TPD结果相一致。

PeakⅠ:Si(4Al,0Si)in SA domain;PeakⅡ:Si(4Al,0Si)in SAPO;PeakⅢ:Si(3Al,1Si)in SA domain;PeakⅣ:Si(2Al,2Si)in SA domain;PeakⅤ:Si(1Al,3Si)in SA domain;PeakⅥ:Si(0Al,4Si)in SA domain图8 Ni/SAPO-11-x催化剂的29Si MAS NMR图谱Fig.8 29Si MAS NMR spectra of the Ni/SAPO-11-x catalysts(a)Ni/SAPO-11-0.2;(b)Ni/SAPO-11-0.4;(c)Ni/SAPO-11-0.6;(d)Ni/SAPO-11-0.8

表4 Ni/SAPO-11-x催化剂中具有不同配位环境硅物种的相对含量Table 4 The relative content of Si with different coordination environments over the Ni/SAPO-11-x catalysts
2.8 催化剂异构化性能评价
以正己烷为模型化合物对Ni/SAPO-11-x催化剂的临氢异构化性能进行评价,结果如图9和图10所示。由图9可以看出,4种催化剂对应的正己烷转化率均随反应温度的升高而增加,且在较低温度范围内(280~340 ℃)增加迅速,随反应温度升高,临氢异构化反应过程逐渐趋于动力学平衡,正己烷转化率上升速率逐渐降低。不同Si含量制备的Ni/SAPO-11-x催化剂作用下对应正己烷的转化率由大到小的顺序依次为:Ni/SAPO-11-0.6、Ni/SAPO-11-0.4、Ni/SAPO-11-0.8≈Ni/SAPO-11-0.2。图6中TEM测试结果表明,初始凝胶中Si的含量并不能显著影响催化剂表面Ni颗粒尺寸,即Ni/SAPO-11-x催化剂表面金属位的数量接近,此时催化剂的异构化活性取决于其酸性质。Ni/SAPO-11-0.6 及Ni/SAPO-11-0.4具有较多的中强酸性位,因此两者的活性较高。

图9 Ni/SAPO-11-x催化下的正己烷转化率随反应温度变化曲线Fig.9 The n-hexane conversion versus reaction temperature for the Ni/SAPO-11-x catalystsReaction conditions:p=2.0 MPa;n(H2)/n(n-Hexane)=4.0;MHSV=1.0 h-1;t=4 h
由图10可知,4种Ni/SAPO-11-x催化剂的异己烷选择性均随正己烷转化率的上升(即随着温度升高)而降低,这归因于高温对正己烷的裂化反应有促进作用。催化剂表面发生的异构化反应与裂化反应为竞争反应,烷烃的异构化为放热反应,因此升高反应温度不利于反应平衡向异构化方向进行,由于异构化反应的活化能低于裂化反应,所以高反应温度在加快异构化反应的同时更利于提高裂化反应的速率,导致异己烷择性降低。在相同正己烷转化率条件下,4种催化剂的异己烷总选择性接近,且在正己烷转化率不高于 75%时,异己烷的总选择性均高于90%,这表明催化剂酸性的改变并未影响催化剂的异构烃选择性。

图10 Ni/SAPO-11-x催化正己烷反应的异己烷选择性Fig.10 The i-hexane selectivity versus n-hexane conversion for the Ni/SAPO-11-x catalystsReaction conditions:p=2.0 MPa;n(H2)/n(n-Hexane)=4.0;MHSV=1.0 h-1;t=4 h
Ni/SAPO-11-x催化剂作用在正己烷临氢异构化反应中时,甲基戊烷(MP)和二甲基丁烷(DMB)的选择性随正己烷转化率变化如图11所示。由图11 可知:由于SAPO-11分子筛的孔道限域效应和较弱的酸性质,正己烷临氢异构化生成的异己烷以MP为主,仅有少量DMB生成。且MP产物中的2-甲基戊烷(2-MP)的动力学尺寸小于3-甲基戊烷(3-MP);DMB中的2,3-二甲基丁烷(2,3-DMB)的动力学尺寸小于2,2-二甲基丁烷(2,2-DMB),2-MP和2,3-DMB的选择性分别高于3-MP和2,2-DMB。在较高正己烷转化率条件下,Ni/SAPO-11-0.8对应的MP选择性相对较低,而DMB选择性较高,这主要归因于该催化剂具有较高的介孔含量,因为孔道尺寸的增大可减弱Ni/SAPO-11-0.8催化剂对尺寸较大的多支链异构体生成的空间限制作用,提高反应中DMB的选择性。烷烃临氢异构化催化剂的反应性能不仅取决于金属位的加氢/脱氢活性和酸性位的骨架异构活性,还与两活性位的协同作用密切相关。当催化剂金属位相对酸性位不足时,发生在金属位的脱氢/加氢反应为整个过程的速控步骤,此时异构烯烃中间体无法及时在金属位表面加氢饱和,其裂化反应机率增大,导致异构烃选择性较低,提高金属位数量则可在提高催化剂活性的同时改善其异构烃选择性;而当金属位过量时,反应的速控步骤为发生在酸性位的骨架异构化反应,此时过量金属位在赋予催化剂高活性的同时,还可使烯烃中间体及时加氢饱和,减少裂化反应的发生,提高异构烃选择性[31]。笔者前期的研究发现[17],Ni/SAPO-11-0.4 催化剂过量的金属位可使得异构化过程中的烯烃中间体快速加氢,降低裂化反应的可能性,提高异构烃选择性,此时提高催化剂的酸量是改善其催化性能的根本途径。相比于Ni/SAPO-11-0.4 催化剂,Ni/SAPO-11-0.6的酸量增加显著提高催化剂的活性,但对异构烃选择性并未产生影响,表明该催化剂表面金属位与酸性位仍可实现高效协同。
将Ni/SAPO-11-0.6催化剂与Pt/SAPO-11催化剂的异构化反应性能进行对比分析,结果如图12所示。由图12可以看出,在相同反应条件下,Ni/SAPO-11-0.6 催化剂对应的正己烷转化率和异己烷收率均高于Pt/SAPO-11催化剂,而两者的异构烃选择性及裂化选择性(sCracking)接近,验证了Ni/SAPO-11-0.6 催化剂具有较高活性的同时,表面金属位与酸性位还可实现高效协同的结论,同时这一结果也表明该催化剂具有替代贵金属异构化催化剂的可能性。

DMB—Dimethyl butane;MP—Methyl hexane图11 Ni/SAPO-11-x催化正己烷临氢异构化反应性能Fig.11 The catalytic performance of Ni/SAPO-11-x catalysts in n-hexane hydroisomerization(a)The selectivity of MP and DMB;(b)The variation curves of isomerized products with n-hexane conversionReaction conditions:p=2.0 MPa;n(H2)/n(n-Hexane)=4.0;MHSV=1.0 h-1;t=4 h

图12 Ni/SAPO-11-0.6及Pt/SAPO-11催化剂的异构化性能Fig.12 Catalytic performance for the Ni/SAPO-11-0.6 and Pt/SAPO-11 catalystsReaction conditions:p=2.0 MPa;T=320 ℃;n(H2)/n(n-Hexane)=4.0;MHSV=1.0 h-1;t=4 h
3 结 论
(1)采用原位干胶法合成Ni/SAPO-11异构化催化剂时,通过改变初始凝胶中Si含量可有效调控Ni/SAPO-11催化剂酸性质,而对其形貌、金属物种类型及分散度无显著影响,该研究结果可为其他双功能催化剂的设计和制备提供理论指导。
(2)增加初始凝胶中Si含量可促进Si同晶取代进入SAPO-11分子筛骨架,提高骨架硅含量,但也会增加硅岛结构的相对含量。
(3)当初始凝胶中SiO2与Al2O3摩尔比为0.6时,得到的Ni/SAPO-11催化剂的酸量较高,该催化剂在正己烷临氢异构化中的活性及异构烃选择性优于Pt/SAPO-11催化剂,具有替代贵金属异构化催化剂的可能性。
猜你喜欢
化工设计(2022年4期)2023-01-02
哈尔滨理工大学学报(2022年1期)2022-05-10
化学研究(2022年1期)2022-02-27
石油炼制与化工(2022年2期)2022-02-15
石油石化绿色低碳(2020年2期)2020-12-31
化工管理(2020年26期)2020-10-09
当代化工(2019年1期)2019-12-12
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
电子技术与软件工程(2016年24期)2017-02-23
