
提高催化重整反应选择性的反应路径设计与研究
2021-12-22 06:55王嘉欣马爱增
石油学报(石油加工) 2021年6期
王嘉欣,马爱增,于 宁
(中国石化 石油化工科学研究院,北京 100083)
催化重整是将石脑油转化为高辛烷值汽油调合组分或芳烃,同时产生大量氢气的重要石油化工过程[1-2]。随着科学技术的不断进步,现有催化重整技术的产品实际收率已经越来越接近其理论收率[3]。催化重整中有利于生成芳烃和高辛烷值汽油调合组分的主要反应,包括六元环烷烃脱氢、五元环烷烃脱氢异构、链烷烃脱氢环化及链烷烃异构化等;副反应包括饱和烃类的氢解和加氢裂化等,生成轻烃产物、少量的芳烃脱烷基和烷基转移等,以及中间产物烯烃的聚合、环化生成稠环化合物导致催化剂表面积炭的生焦反应。各类主要重整反应的热力学和动力学特性如表1所示[3]。
表1中前3个反应都可以达到热力学平衡,后2类反应则受动力学控制。最有利于提高辛烷值和芳烃产率的反应,如六元环烷烃脱氢、五元环烷烃脱氢异构和烷烃的脱氢环化反应,低压和高温条件均利于反应进行。其中烷烃脱氢环化反应的速率相当慢,而这类反应在重整所有反应中对辛烷值的贡献最为明显;同时,由于脱氢环化时1 mol烃可以产生4 mol氢气,也是重要的产氢反应。然而,烷烃脱氢环化反应进行的同时,总伴随着烷烃加氢裂化反应。在工业生产中,当反应压力和空速固定时,提高芳烃产率的同时往往伴随着液体收率的损失,这是影响催化重整反应选择性最主要的一对矛盾。

表1 各类重整反应热力学和动力学的比较Table 1 Thermodynamic and kinetic comparison of the main reforming reactions
因此,寻找新催化材料打破现有技术路线的限制,进一步提高催化重整反应的汽油收率或芳烃收率,成为催化重整技术进步的研究重点。笔者在分析现有重整反应路径的基础上,通过分子模拟对正庚烷脱氢环化过程中的决速步骤反应能垒进行理论分析,提出降低决速步骤反应能垒的可行途径,设计出提高重整反应选择性的反应路径,制备了含硼硅分子筛的催化剂,实现了催化重整芳烃和液体选择性不同程度的提高,并以不同模型化合物为原料,通过分析反应产物分布,验证了新反应路径的可行性。
1 实验部分
1.1 分子模拟计算
利用Material studio 6.0构建反应物及催化剂的结构模型,利用基于泛函理论的DMol 3模块进行几何结构优化并计算体系的自由能、C—C和C—H键的裂解能及基元反应的反应能垒。在DMol 3计算中,选用泛函GGA-PW91方法,收敛精度为:能量0.05 kJ/mol,受力1×1012N,位移5×10-13m。利用Adsorption locator模块计算反应物、中间态及产物在催化剂表面的吸附状态,力场采用COMPASS,电荷使用Forcefield,精确度为Fine。利用Morphology模块计算催化剂的最大可见晶面,力场采用COMPASS,电荷使用Forcefield assigned,精确度为Medium。
1.2 原料和试剂
正庚烷,分析纯,北京化工厂产品;1-己烯,分析纯,百灵威试剂公司产品;环己烷,分析纯,北京益利精细化学品有限公司产品;Pt(NH3)4Cl2,北京佳友盛新技术开发中心产品。
硅铝β分子筛(以下简称Aβ)、硼硅β分子筛(以下简称Bβ)、PtRe/Al2O3半再生重整催化剂(工业牌号PR-D)、PtSn/Al2O3连续重整催化剂(工业牌号PS-VI),均来自中国石化催化剂公司。
1.3 催化剂制备
将一定量的Pt(NH3)4Cl2和去离子水配制为浸渍液,按照液/固体积比4∶1把浸渍液分别加入至Bβ分子筛和Aβ分子筛粉体中,室温搅拌4 h,静置分层,移去上层清液并于120 ℃下干燥12 h,然后在300 ℃下焙烧2 h后制得Pt质量分数均为1%的Pt/Bβ催化剂和Pt/Aβ催化剂,粉体催化剂经压片成型后破碎为40~60目的颗粒。采用机械混合的方式将粒径40~60目的不同种催化剂按一定的体积比混合均匀,制成用于催化剂反应性能评价的混合催化剂。
1.4 催化剂表征
采用Bruker公司生产的IFS113Ⅴ型傅里叶变换红外光谱仪进行吡啶吸附红外分析。采用Horiba公司生产的EMIA-820Ⅴ型碳硫分析仪进行反应后催化剂积炭含量分析。
1.5 催化剂评价
催化剂的反应性能评价在固定床微型反应装置上进行。固定床微型反应装置主要由进料器、反应器、在线采样分析和产品收集4部分组成,反应器为内径1 cm的不锈钢反应器。将1 mL粒径为40~60目的催化剂填装于反应器恒温段,为减少热效应,用相同目数的细石英砂按体积比4∶1稀释催化剂。采用质量流量计控制氢气流量,微量泵控制原料油流量,使其按所需比例混合后进入反应器。催化剂在氢气气氛下升温至480 ℃还原2 h,然后降温至370 ℃,同时注入反应原料,流量稳定后升温至反应温度,稳定1 h后开始在线采样分析。产物组成采用日本岛津公司生产的GC-14C型气相色谱仪在线检测,测试条件为:载气N2,FID检测器,PONA色谱柱。原料的转化率(x,%)和产物产率(y,%)采用面积归一法计算:
x=[1-Ar×Br/∑(Ai×Bi)]×100%
(1)
y=(Aj×Bj)/∑(Ai×Bi)×100%
(2)
式(1)和式(2)中:Ai—i组分色谱峰面积;Bi—i组分相对质量校正因子;r—原料;j—目标产物。
2 结果与讨论
2.1 分子模拟计算
提高催化重整反应选择性的关键问题在于,提高烷烃脱氢环化选择性的同时抑制裂化反应。分子模拟以正庚烷为初始反应物,通过对比需要克服的反应能垒,计算得出优先生成的中间产物,结合反应物在催化剂表面的吸附、扩散等因素,综合推论正庚烷脱氢环化生成甲苯的最优反应路径。正庚烷脱氢环化反应路径包括:烷烃脱氢生成烯烃(见式(3))、烯烃脱除氢负离子(见式(4))、碳正离子移位(见式(5))、烯烃碳正离子的环化(见式(6))和扩环(见式(7))、氢负离子转移(见式(8))、甲基环己烷脱氢生成甲苯(见式(9)、(10))等基元反应。整个反应路径中的速控步骤为正庚烯在L酸中心脱除氢负离子生成烯烃碳正离子(见式(4)),需要克服的反应能垒最高为244.86 kJ/mol。

(3)

(4)

(5)

Ea=59.05 kJ/mol
(6)

Ea=162.57 kJ/mol
(7)
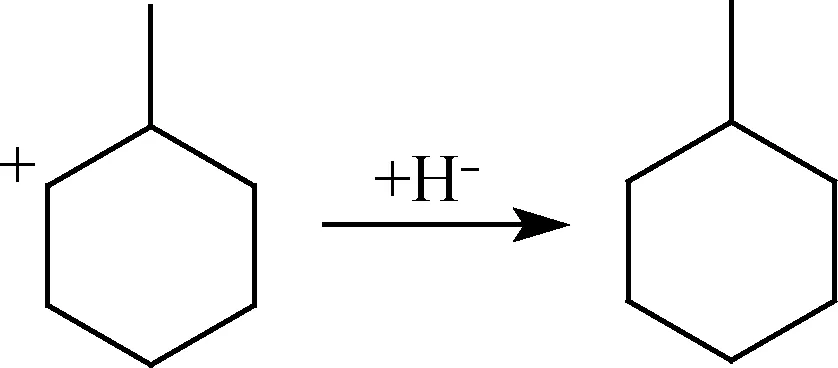
Ea=15.14 kJ/mol
(8)

Ea=79.02 kJ/mol
(9)

Ea=56.94 kJ/mol
(10)
为了降低烷烃脱氢环化反应路径中的最高反应能垒,需要优先解决正庚烯在L酸中心脱除氢负离子生成烯烃碳正离子反应能垒过高的问题。可行的操作手段包括,调变现有氧化铝载体的L酸强度,或者引入适当的B酸。在氧化铝载体的酸性调变方面已经进行了大量的工作,进一步改进的空间不大。因此,研究过程中主要考虑催化剂体系中引入B酸中心对氧化铝重整催化剂正庚烷脱氢环化反应路径的影响。分子模拟计算结果表明,反应中间产物正庚烯极易与氢质子反应生成正庚烷碳正离子(见式(11)),需要克服的反应能垒仅为4.40 kJ/mol。正庚烷碳正离子与正庚烯发生双分子间氢负离子转移反应生成庚烯碳正离子(见式(12))需要克服的反应能垒为36.70 kJ/mol,远低于式(4)反应的能垒。从理论上分析,促进双分子间氢负离子转移反应将是解决生成烯烃碳正离子反应能垒过高问题的可行方案。

(11)

Ea=36.70 kJ/mol
(12)
下面以正庚烷脱氢环化反应路径中的速控步骤——正庚烯在L酸中心脱除氢负离子反应(式(4))和正庚烷碳正离子与正庚烯发生双分子间氢负离子转移反应(式(12))为例,进行模型构建和计算。
2.1.1 正庚烯脱除氢负离子基元反应的模拟过程
选用AlCl3分子作为L酸活性中心,模拟计算了2-庚烯在L酸活性中心发生脱除氢负离子的基元反应,反应结构示意图如图1所示。从图1可以看出,sp2杂化的Al原子具有吸取1对电子的能力,可以脱除2-庚烯中氢负离子(H-)。

图1 脱除氢负离子反应的结构示意图Fig.1 Schematic diagram of molecular structures of hydrogen anion removal reaction(a)Reactant;(b)Transition state;(c)Product
2.1.2 双分子间氢负离子转移基元反应的模拟过程
选用氢正离子(H+)作为活性中心,模拟计算了2-庚烯和庚烷-2-碳正离子发生双分子间氢负离子转移的基元反应。图2为反应结构示意图,图中黄色球体代表碳正离子。从图2可以看出,庚烷-2-碳正离子中的碳正离子可以吸取2-庚烯中烯丙位富含电子的氢负离子,生成容易发生环化反应的烯烃碳正离子。

图2 双分子间氢负离子转移反应的结构示意图Fig.2 Schematic diagram of molecular structures of bimolecular hydride transfer reaction(a)Reactant;(b)Transition state;(c)Product
2.2 反应路径设计
如上所述,正庚烷脱氢环化路线中的速控步骤为正庚烯在L酸中心脱除氢负离子生成烯烃碳正离子的反应,需要克服的反应能垒为244.86 kJ/mol;而当反应引入氢质子后,正庚烯优先与氢质子反应生成庚烷碳正离子,随后,庚烷碳正离子与正庚烯发生双分子间氢负离子转移反应,生成庚烯碳正离子,需要克服的反应能垒为36.70 kJ/mol。庚烯碳正离子的生成不再受高反应能垒的限制,降低了正庚烷脱氢环化路线中的最高反应能垒。据此提出“通过增强重整反应网络中氢负离子转移反应活性,提高催化重整反应选择性”的反应路径设计。
前期分子筛在催化重整中应用的大量研究结果表明,对于B酸的引入,常规硅铝分子筛的强酸性导致裂化反应严重,影响芳烃产率和液体收率[4-5]。因此,需要寻找或开发酸强度适宜的新型催化材料。硼硅分子筛的酸强度明显低于硅铝分子筛[6-7],而且硼进入分子筛骨架能调变分子筛的表面酸强度[8]。笔者考察了硼硅β分子筛的重整反应性能,并以不同模型化合物为原料对上述设计的反应路径进行验证。
2.3 硼硅β分子筛酸性与催化性能
用吡啶吸附红外光谱法对γ-Al2O3,硅铝β分子筛(Aβ)和硼硅β分子筛(Bβ)的表面酸类型和酸强度进行表征,1540 cm-1附近的吸收峰为B酸,1450 cm-1附近的吸收峰为L酸,通过不同吸收峰的面积计算出相应的酸量,不同载体的表面酸性质见表2。由表2可知,γ-Al2O3没有检测到B酸中心。硼硅β分子筛的酸量明显弱于硅铝β分子筛,特别是B酸酸量。

表2 不同催化剂载体的酸性质Table 2 Acid properties of different catalyst supports Acid amount/(μmol·g-1)
分别以Aβ分子筛和Bβ分子筛为载体,负载金属铂后制得Pt/Aβ和Pt/Bβ催化剂,以正庚烷为原料,在500 ℃、体积空速6 h-1、压力1 MPa和氢/油体积比1200条件下,考察催化剂反应性能,并与工业氧化铝重整催化剂PR-D对比,实验结果如表3所示。由表3可知,随着分子筛载体酸性的减弱,催化剂所对应反应的气体产率降低,芳烃产率增加。由于硅铝分子筛的强酸性,导致Pt/Aβ催化剂对应反应中正庚烷完全裂解为气体产物;Pt/Bβ催化剂对应反应中的气体产率明显低于Pt/Aβ催化剂,但仍远高于PR-D催化剂。具有较强裂解性能的Pt/Bβ催化剂和混合催化剂对正庚烷重整反应性能的结果如表4所示。由表4可知:与PR-D催化剂相比,当Pt/Bβ催化剂与PR-D催化剂混合后,对应反应的气体产率不但没有增加,反而降低,同时芳烃选择性和芳烃产率也较二者单独使用时明显增加。Pt/Bβ催化剂与PR-D催化剂等体积比例混合使用时,对应的反应具有最低的气体产率和较高的芳烃产率。

表3 不同载体催化剂对正庚烷重整反应性能的影响Table 3 Effects of supports on heptane reforming reaction performance over different catalysts

表4 Pt/Bβ、PR-D及其机械混合催化剂对正庚烷重整反应性能的影响Table 4 Performance of heptane reforming reaction over Pt/Bβ and PR-D catalyst mixtures
2.4 混合催化剂反应机理探究
上述实验结果表明,重整催化剂体系引入弱B酸中心后,确实增加了正庚烷脱氢环化反应的选择性。但需要进一步证实是否是通过增强氢负离子转移反应路径实现的。按照以往的认知,相对于氧化铝催化剂,酸性较强的硼硅分子筛催化剂加入到催化重整反应体系中,应促进裂解反应的发生。然而,实验结果表明(表4),引入一定比例的硼硅分子筛催化剂不仅增加了催化重整反应的芳烃产率,而且气体产率同时降低。推测产生这一结果的可能原因有以下3种。
(1)反应空速的影响
对于混合催化剂当中的PR-D催化剂,分子筛催化剂的加入相当于增加了反应空速,高空速引起产物气体产率降低、芳烃产率增加。
(2)轻烃芳构化反应
分子筛催化剂将裂解产物通过轻烃芳构化二次反应生成芳烃。
(3)不同的反应路径
2.4.1 反应空速的影响
在反应温度500 ℃、反应压力1.0 MPa、氢/油体积比1200的条件下,通过将PR-D催化剂与惰性石英砂混合提高PR-D催化剂的反应空速,以正庚烷为原料考察了空速对反应结果的影响,结果列于表5。从表5可以看出,在高空速下,产物的气体产率降低,但同时芳烃产率下降,因此空速影响不是混合催化剂协同作用产生的原因。

表5 反应空速对Pt/Bβ与PR-D催化剂正庚烷重整反应性能的影响Table 5 Effects of LHSV on heptane reforming reaction performance over Pt/Bβ and PR-D catalysts
2.4.2 轻烃芳构化反应
C3、C4轻烃在酸性分子筛上可以经芳构化反应生成包含苯、甲苯、二甲苯、乙苯及更高碳数的芳烃混合物[9]。据轻烃芳构化机理,如果酸性较强的Bβ分子筛促进反应过程产生的气体产物发生轻烃芳构化二次反应,芳烃产物分布中应含有C6~C9多种芳烃组分。然而,实际的芳烃产物中只有苯和甲苯。表6中列出了PR-D和混合催化剂所对应反应的气体产物组成。由表6中的产物分布可知,混合催化剂对应反应的气体产物中C3和C4烷烃的比例没有明显降低。同时,以典型的轻烃芳构化催化剂Zn、P改性的ZSM-5分子筛与PR-D复配催化下轻烃芳构化反应几乎全部生成裂化产物。因此,轻烃芳构化二次反应不是混合催化剂对应反应中气体产率低、芳烃产率增加的主要因素。

表6 PR-D和混合催化剂对正庚烷重整反应气体产物组成的影响Table 6 Composition of gas products over PR-D catalyst and catalyst mixtures with Pt/Bβ
2.4.3 氢负离子转移反应
(1)正庚烷在N2气氛下的氢负离子转移反应考察
大量的研究表明[10-12],链烷烃脱氢环化过程首先在金属中心上脱氢生成烯烃,然后在酸性中心上异构、环化,进一步在金属中心上脱氢生成芳烃。烯烃或碳正离子是脱氢环化的中间产物,同时也是双分子氢负离子转移反应的反应物,但在重整临氢的反应条件下,产物中的烯烃极易饱和,不能像常规氢转移反应以烯烃和烷烃比例来衡量氢负离子转移活性。因此,为了考察重整反应体系中是否存在氢负离子转移反应,比较了非临氢条件下PR-D和混合催化剂上芳烃和烯烃生成。对于氢负离子转移反应活性较强的催化剂,作为中间产物的烯烃将更多地被转化为芳烃,最终产物中的烯烃含量较少。
实验选取正庚烷为模型化物,在N2气氛下对催化剂反应性能进行了评价,结果如表7所示。由表7可知,与PR-D单独使用相比,混合催化剂体系对应反应产物中芳烃含量高而烯烃含量较少,说明分子筛催化剂的引入提高了氢负离子转移反应的活性。

表7 N2气氛下不同催化剂对应正庚烷重整反应的结果Table 7 Results of heptane conversion over different catalysts under N2 atmosphere
(2)己烯与环己烷混合原料氢负离子转移反应实验
环烷烃失去氢负离子后形成稳定的正碳离子,是好的潜在供氢体,而烯烃在质子化后是很强的负氢离子接受体[13],这2类烃是验证是否发生氢负离子转移反应的适宜模型化合物。以环己烷质量分数为15%的环己烷和1-己烯混合物为反应物,以N2为载气与反应物混合进料,在固定床微型反应器上对催化剂进行评价,实验条件为:温度500 ℃、空速6.0 h-1、反应压力1.0 MPa、气/油体积比1200。反应结果如表8所示。
1-己烯在H2气氛下可以通过金属中心直接加氢饱和,而在N2气氛下,1-己烯的饱和也可以通过双分子氢负离子转移反应进行,因此通过比较不同催化剂体系下烯烃的转化率以及饱和烃的生成量,可以验证混合催化剂体系是否促进了氢负离子转移反应的发生。从表8可以看出:PR-D催化剂单独使用时,1-己烯的转化率较低,同时产物中生成的己烷量也较少;而引入分子筛的混合催化剂上,1-己烯转化率明显增加,产物中己烷和苯的生成量也较高。说明分子筛催化剂的引入,促进了氢负离子转移反应的发生。

表8 环己烷和1-己烯混合原料在不同催化剂上的反应结果Table 8 Results of cyclohexane and 1-hexene conversion over different catalysts
2.5 混合催化剂作用下的氢负离子转移反应机理
基于上述研究结果,混合催化剂体系表现出更高的氢负离子转移活性。氢负离子转移反应不仅可以促进烯烃转化为芳烃,而且可以终止裂化反应[14]。以正庚烷为模型化合物,推测混合催化剂协同作用产生的可能机理如图3所示。由图3可知:PR-D催化剂具有好的烷烃脱氢活性,正庚烷首先在PR-D催化剂的金属中心上脱氢生成烯烃,烯烃可以通过脱除氢负离子形成烯烃碳正离子,然后经环化、脱氢生成芳烃;此外,烯烃在酸中心上形成碳正离子,进一步通过裂化反应生成气体小分子。其中,生成烯烃碳正离子是生成芳烃的关键中间物种。在混合催化剂体系中,Bβ分子筛催化剂的引入,提供了有利于烯烃生成碳正离子的B酸中心,而且B酸中心可以促进烯烃和碳正离子发生氢负离子转移反应,生成有利于芳构化反应的烯烃碳正离子,同时使裂化反应的前生物碳正离子饱和生成烷烃,从而在抑制裂解反应的同时促进了芳烃的生成。
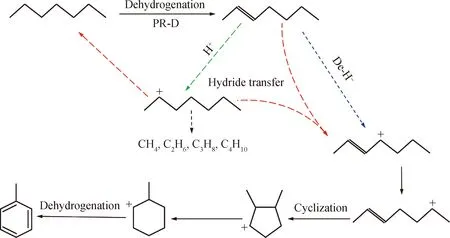
图3 混合催化剂作用下的氢负离子转移反应路径Fig.3 Reaction routes of hydride transfer over mixed catalysts
2.6 混合催化剂对石脑油催化重整反应性能的研究
以石脑油为原料在中型试验装置上考察了工业半再生重整催化剂PR-D和其与Bβ分子筛催化剂机械混合催化剂3PRZ的催化重整反应性能。3PRZ催化剂由体积分数分别为30%的Pt//Bβ催化剂和70%的PR-D催化剂机械混合制得。石脑油原料组成如表9所示。
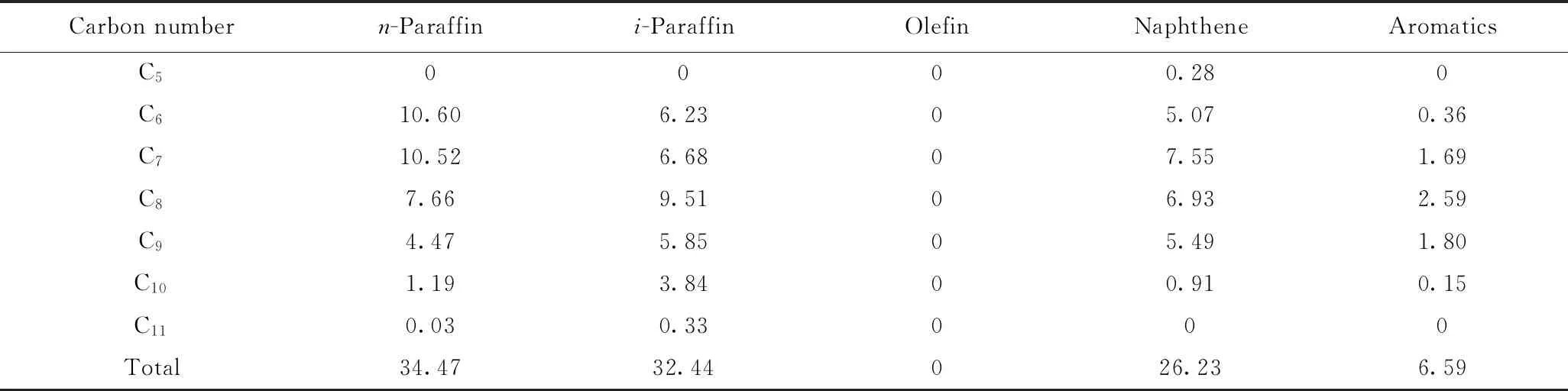
表9 石脑油族组成Table 9 Composition of naphtha feed w/%
在反应温度500~540 ℃、压力1.0 MPa、体积空速1.8 h-1和氢/油体积比1000的反应条件下,混合催化剂3PRZ和工业氧化铝重整催化剂PR-D对石脑油催化重整反应结果如图4所示。以石脑油为反应原料时,比较液体产品中芳烃含量与液体产品收率的关系可以衡量催化剂的反应选择性。由图4可知,与PR-D催化剂相比,在液体产品中芳烃含量相同的条件下,3PRZ催化剂作用反应的液体产品收率更高,说明混合催化剂具有更好的选择性。
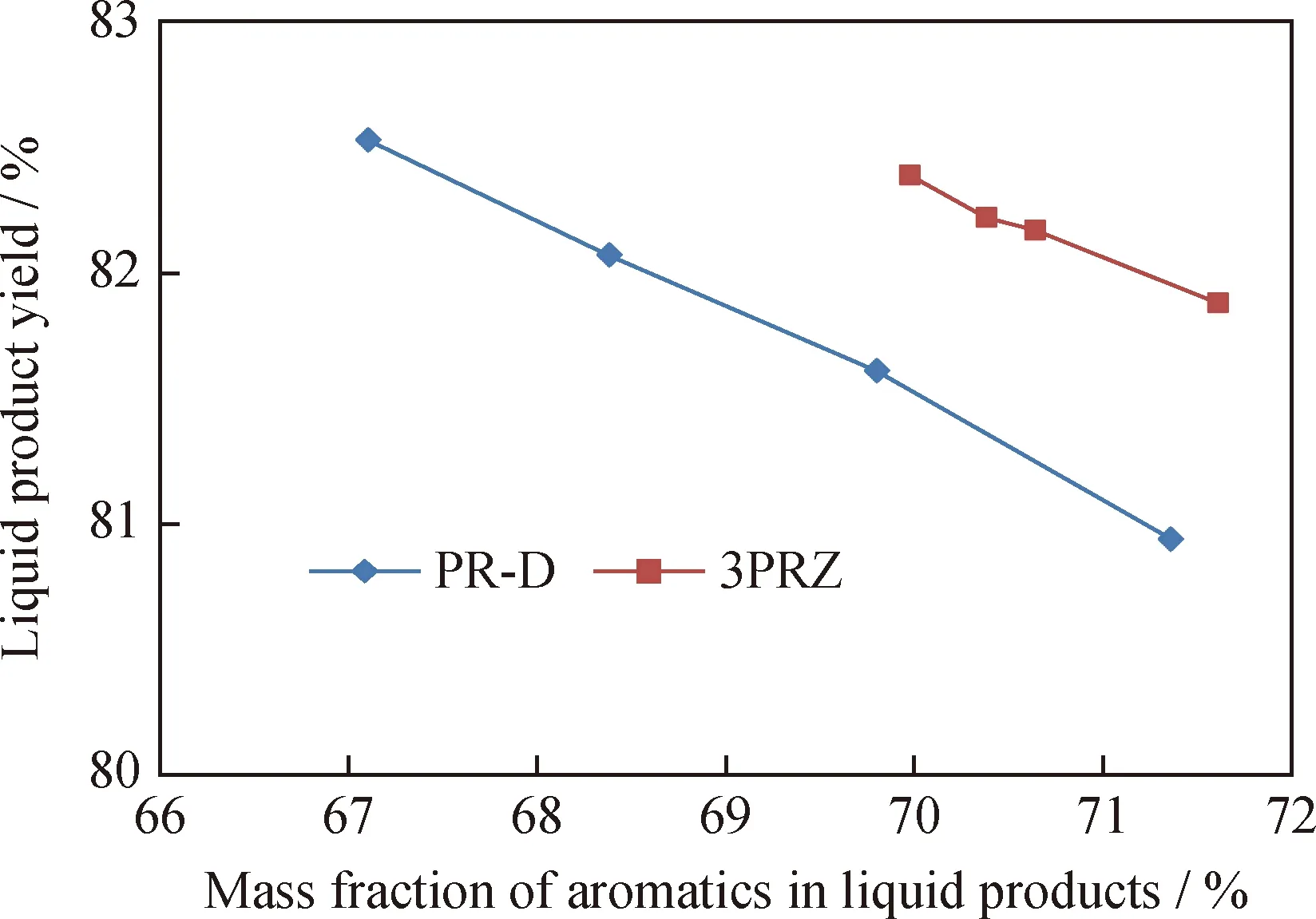
图4 3PRZ和PR-D催化剂液体产品收率和液体产品中芳烃质量分数的关系Fig.4 Relationship between liquid products yield and aromatics mass fraction in liquid products over 3PRZ and PR-D catalystsLHSV:1.8 h-1;Volume ratio of H2 and oil:1000;Pressure:1.0 MPa;Temperature:500—540 ℃;Catalyst volume:50 mL
在压力1.0 MPa、体积空速1.8 h-1和氢/油体积比1000的反应条件下,考察混合催化剂3PRZ和PR-D催化剂对石脑油催化重整反应的稳定性,先在中型试验装置上520 ℃下反应232 h,然后升温至540 ℃继续反应,反应结果如图5所示。由图5可知,与PR-D催化剂相比,3PRZ催化剂对石脑油催化重整反应具有更好的稳定性和更高的芳烃产率,特别是在540 ℃下,3PRZ催化剂对石脑油催化重整反应的稳定性明显好于PR-D催化剂。在反应时间330 h的试验后期,3PRZ催化剂对应的芳烃质量收率比PR-D催化剂高7.3%。对反应后催化剂的积炭含量进行分析,3PRZ催化剂的积炭速率比PR-D催化剂低6%。由此可见,当按照促进氢负离子转移反应路径设计的催化剂体系用于石脑油原料催化重整时,同样可以提高该反应的选择性,而且还能降低积炭速率。

图5 3PRZ和PRT-D催化剂石脑油反应稳定性试验结果Fig.5 Results of stability test with naphtha feed over 3PRZ and PR-D catalystsLHSV:1.8 h-1;Volume ratio of H2 and oil:1000;Pressure:1.0 MPa;Catalyst volume:50 mL
2.7 硼硅β分子筛对连续重整催化剂的促进作用
以正庚烷为原料,在500 ℃、体积空速10 h-1、压力0.7 MPa和氢/油体积比1000条件下,考察连续重整催化剂PS-Ⅵ、PS-Ⅵ与Pt/Bβ的混合催化剂、Pt/Bβ催化剂的反应性能,结果如表10所示。由表10可知,与半再生催化剂类似,Bβ分子筛与PS-Ⅵ连续重整催化剂混合使用时,可以提高重整反应的选择性,获得更高的芳烃产率和更低的气体产率。因此,促进氢负离子转移反应路径的设计对于PtRe和PtSn重整催化剂体系均具有适用性。

表10 Pt/Bβ与PS-Ⅵ机械混合催化剂作用于正庚烷重整反应的性能Table 10 Performance of heptane reforming reaction over Pt/Bβ and PS-Ⅵ catalyst mixtures
3 结 论
在理论分析和分子模拟的基础上,设计出“通过增强重整反应网络中氢负离子转移反应活性,提高催化重整反应选择性”的新反应路径,以杂原子硼硅β分子筛与氧化铝重整催化剂混合使用,将弱B酸中心引入到重整催化剂反应体系中,促进了重整反应网络中氢负离子转移反应活性,提高了重整反应选择性,获得了更高的芳烃产率和液体收率。促进氢负离子转移的反应路径的设计,对于半再生重整和连续重整催化剂体系均具有适应性,对目的产品产率最大化具有实际应用意义,为新型重整催化剂的开发提供了研究思路。
猜你喜欢
上海建材(2022年3期)2022-11-04
工程技术与管理(2022年7期)2022-03-04
浙江林业科技(2022年1期)2022-02-20
科学与生活(2021年3期)2021-11-10
石油炼制与化工(2021年3期)2021-03-23
哈尔滨工业大学学报(2020年1期)2020-12-21
化工时刊(2020年11期)2020-01-12
中国特种设备安全(2019年9期)2019-12-03
现代农村科技(2018年5期)2018-05-31
郑州大学学报(工学版)(2014年6期)2014-03-01
