
二甲双胍对高脂喂养糖尿病大鼠心肌细胞PGC-1α及PI3K表达的影响
2021-12-17 02:32张颖,徐静,李楠
山西医科大学学报 2021年11期
张 颖,徐 静,李 楠
(1西安交通大学第二附属医院内分泌科,西安 710032;2空军军医大学第一附属医院内分泌科;3西安市中心医院内分泌科;*通讯作者,E-mail:xujingjdey85@163.com)
高游离脂肪酸血症和高甘油三酯血症可导致心肌脂质沉积,心肌细胞脂肪酸氧化代谢增强可直接引起心肌细胞的耗氧量增高,长脂酰辅酶A的增多,诱导细胞内超氧化簇的聚集、抑制一氧化氮合酶活性及引起心肌细胞凋亡,进而引起心室壁增厚、心室重构、从而引起心脏收缩功能障碍等。糖尿病心肌对脂肪酸的摄取和利用远大于心肌细胞实际的氧化代谢需要,脂肪酸的过度活化可诱发心肌脂毒性,导致心肌细胞脂代谢产物和介质堆积,线粒体活性氧产生过量,进而引起糖尿病心肌肥大和功能障碍[1]。因此,脂毒性与心肌损害的发生发展密切相关。胰岛素抵抗(insulin resistance,IR)伴发的3个特征性代谢紊乱(高血糖症、高胰岛素血症、高脂血症)也可通过各种途径导致心肌间质纤维化的发生,与心肌能量代谢、心肌细胞胰岛素敏感性亦密切相关。二甲双胍对心血管有一定的保护作用,已被较多的循证医学证据支持[2,3],但二甲双胍对心肌作用机制仍需探讨。过氧化物酶体增殖物激活受体γ协同刺激因子-1α(peroxisome proliferator activated receptor γ co-activator 1α,PGC-1α)作为心肌线粒体能量代谢的主要因子[4],PGC-1α在肝脏中的表达可被二甲双胍所抑制,在肝脏中,二甲双胍可抑制由于PGC-1α过表达导致的磷酸烯醇式丙酮酸脱羧激酶(phosphoenolpyruvate carboxykinase,PEPCK)以及6-磷酸葡萄糖脱氢酶(glucose-6-phosphate dehydrogense,G-6-Pase)增加[5],但二甲双胍并不影响PGC-1α介导的线粒体基因诱导。目前二甲双胍对心肌细胞PGC-1α作用尚不明确。该研究通过观察二甲双胍对高脂喂养并且由链脲佐菌素(streptozotocin,STZ)诱导的2型糖尿病(T2DM)大鼠模型心肌细胞PGC-1α及磷酸肌醇3激酶(phosphatidylinositide 3-kinase,PI3K)p85亚基在大鼠心肌细胞表达的影响,以进一步探究二甲双胍对糖尿病大鼠心肌细胞中胰岛素信号传导及心肌细胞能量代谢的作用。
1 材料与方法
1.1 实验动物
选用西安交通大学医学院实验动物中心健康雄性SD大鼠36只,清洁级,体质量180-280 g,血糖正常。饲养于标准环境温度(23±2)℃、湿度(55±5)%,光照维持昼夜循环每日12 h,自由进饮食,每天换水、垫料。
1.2 动物分组及T2DM大鼠模型建立
雄性SD大鼠36只随机分为正常对照组(12只)和2型糖尿病模型组(24只)。正常对照组喂以普通饲料,2型糖尿病模型组喂以高脂高糖饲料(基础饲料82%,猪油10%,胆固醇2.5%,蔗糖5%,胆酸钠0.5%)[6],其中胆固醇、胆酸钠购于上海如吉生物科技发展有限公司。普通饲料购于北京科澳协力饲料有限公司。猪油、蔗糖为自备。喂养至12周末,隔夜空腹给予2型糖尿病模型组大鼠腹腔注射STZ 30 mg/kg(购于美国Sigma公司,于使用前用0.1 mmol/L pH为4.2的柠檬酸-柠檬酸钠缓冲液临时配成1%的溶液)。正常对照组注射相应体积的柠檬酸缓冲液。于实验第15周正常对照组及2型糖尿病模型组进行眼底静脉取血,测空腹血糖(FBG)和空腹胰岛素(FINS)。连续两次以上FBG≥7.8 mmol/L,计算HOMA-IR指数较正常对照组升高有统计学差异者,定义为伴有胰岛素抵抗[7],为造模成功,造模成功大鼠共24只。随机抽取其中12只为糖尿病组,余12只大鼠为干预组。正常对照组和糖尿病组给予等体积生理盐水灌胃。二甲双胍干预组给予盐酸二甲双胍300 mg/(kg·d)灌胃[8],其中盐酸二甲双胍由中美上海施贵宝制药有限公司提供,共持续8周。22周末大鼠禁食水12 h后,称重,用10%的水合氯醛按0.6 g/kg体质量腹腔麻醉,收集血液以测定空腹血糖(FBG)、总胆固醇(TC)、甘油三酯(TG)、高密度脂蛋白(HDL-C)、低密度脂蛋白(LDL-C)、极低密度脂蛋白(VLDL)、游离脂肪酸(FFAs)、空腹胰岛素(FINS),计算胰岛素抵抗指数(HOMA-IR)。留取部分心肌组织固定于10%甲醛,供HE染色,另一部分置于-80 ℃冰箱中,供免疫印迹法检测使用。
1.3 生化指标测定
于22周末进行生化指标测定,血糖采用罗氏血糖仪测定。总胆固醇、甘油三酯、高密度脂蛋白、低密度脂蛋白、极低密度脂蛋白均按试剂盒方法操作。游离脂肪酸采用比色法测定。胆固醇、胆酸钠购于上海如吉生物科技发展有限公司。生物试剂盒购自南京建成生物工程研究所,游离脂肪酸试剂盒购自英国RANDOX公司。胰岛素采用酶联免疫法测定,鼠胰岛素检测试剂盒购自美国R&D公司。
1.4 HE染色
经固定、脱水、制作蜡块、切片脱蜡、染色过程,于光镜下放大至高倍,照相。
1.5 免疫印迹法(Western blot法)检测蛋白表达
心肌组织总蛋白提取后,取上清液分装于离心管中并置于-20 ℃保存。将样品放置于生物分光光度计(NanoDrop ND-1000)比色分析,计算含50 μg蛋白的溶液体积即为上样量。取出上样样品至0.5 ml离心管中,加入5×SDS上样缓冲液与蛋白样品按1 ∶4混匀。SDS-PAGE电泳后转膜,用TBST将一抗稀释至适当浓度,放入1.5 ml离心管中;撕下适当大小的一块儿保鲜膜铺于实验台面上,将抗体溶液加到保鲜膜上;其中PI3K p85多克隆抗体购自美国SCT生物技术公司。PGC-1α多克隆抗体购自英国Abcam生物技术公司。从封闭液中取出膜,将膜蛋白面朝下放于抗体液面上,掀动膜四角赶出残留气泡;放入冰箱内4 ℃孵育过夜。第2天取出膜,用TBST在室温下脱色摇床上洗两次,每次10 min;再用TBS洗一次,10 min。室温下封闭1 h。4 ℃封闭过夜。凝胶成像系统拍照,进行密度值测定。
1.6 统计学处理

2 结果
2.1 大鼠的一般情况
正常对照组12只大鼠普通饲料喂养,毛发光洁、精神状态良好、生长发育正常;2型糖尿病模型组大鼠高脂高糖喂养过程中,毛发易脱落、竖立无光泽,反应迟钝、精神萎靡、生长发育迟缓。第15周用STZ诱导糖尿病大鼠模型成功后,2型糖尿病模型组24只大鼠在原有特殊症状的基础上,出现了多饮、多食、多尿、腹泻、消瘦症状明显。第15周至22周末进行药物干预过程中,正常对照组和糖尿病组仅使用生理盐水灌胃,各自特征较前无特殊改变,其中糖尿病组2只于20周自然死亡;二甲双胍干预组大鼠腹泻较其他组严重,余多饮、多尿症状好转,其中2只于21周因灌胃死亡。
2.2 血糖、血脂、游离脂肪酸、胰岛素及胰岛素抵抗指数的比较
实验室第15周测空腹血糖(FBG)和空腹胰岛素(FINS),连续两次以上FBG≥7.8 mmol/L,HOMA-IR指数较正常对照组升高有统计学差异者定义为伴有胰岛素抵抗,为造模成功(见表1)。22周末测糖尿病组大鼠FBG、TC、TG、LDL-C、VLDL、FFAs、FINS、HOMA-IR指标均较正常对照组大鼠升高(P<0.01),HDL-C指标较正常对照组降低(P<0.01);干预组大鼠FBG、TC、TG、LDL-C、VLDL、FFAs、FINS、HOMA-IR指标均较糖尿病组降低,HDL-C指标较糖尿病组升高,差异均有统计学意义(P<0.01,见表2)。
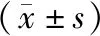
表1 第15周两组大鼠HOMA-IR指标的比较Table 1 Comparison of HOMA-IR of rats between two groups at week
表2 22周末各组大鼠生化指标的比较Table 2 Comparison of the biochemical index of rats between groups at the end of 22
2.3 免疫印迹法结果
与正常对照组比较,糖尿病组PGC-1α表达升高(P<0.05);与糖尿病组比较,干预组PGC-1α表达降低(P<0.05)。与正常对照组比较,糖尿病组PI3K p85表达降低(P<0.05);与糖尿病组比较,干预组PI3K p85表达升高(P<0.05,见图1)。
2.4 HE染色结果
22周末取大鼠心肌组织固定于10%甲醛,行HE染色。正常对照组心肌细胞排列整齐、致密,结构清晰,细胞核形态规则。糖尿病组,可见心肌细胞排列不整齐,心肌细胞肿胀明显,部分细胞核可见形态不规则;心肌细胞部分融合,边界不清楚。二甲双胍干预组,心肌细胞形态排列较整齐,未见明显细胞肿胀,核形态较规则(见图2)。
与正常对照组比较,*P<0.05;与糖尿病组比较,△P<0.05图1 各组心肌组织PI3K p85、PGC-1α表达免疫印迹法结果Figure 1 Western blotting results of PI3K p85 and PGC-1α expression in myocardial tissues in each group

图2 22周末各组心肌组织HE染色结果Figure 2 HE staining results of myocardial tissue in each group at the end of 22 weeks
3 讨论
3.1 脂毒性心肌病与胰岛素抵抗
脂肪酸的摄取和利用不相平衡,造成脂质氧化不充分,不能充分氧化的脂质在心脏沉积,导致心肌功能障碍或心肌肥厚,进而形成脂毒性心肌病[9]。糖尿病患者血浆中FFAs和TAGs水平升高和心肌细胞脂质积聚均为糖尿病性心肌病发展的必然经过。心肌细胞胰岛素抵抗是心肌病发生的一个重要原因。胰岛素抵抗使心肌细胞利用葡萄糖氧化来提供的能量减少,从利用葡萄糖和脂肪酸供能转变几乎全部通过脂肪酸β-氧化提供能量[10]。糖尿病患者心脏长期处于高血糖、高血脂的代谢环境,促进大量基因序列的改变,主要涉及有关免疫反应、细胞信号通路、增殖及转录的基因,继而引起一系列细胞结构和功能的变化[11]。因此,脂毒性与胰岛素抵抗始终互为因果。本实验是在首先诱导动物出现胰岛素抵抗的基础上再注射小剂量STZ,两者结合起来更加有利于诱导出病理、生理方面改变都接近于人类的2型糖尿病,实验中大鼠经高脂高糖喂养后,出现了明显的高脂血症,模型组大鼠胆固醇、甘油三酯、低密度脂蛋白、极低密度脂蛋白及游离脂肪酸、胰岛素抵抗指数均出现明显升高,高密度脂蛋白明显降低,是研究2型糖尿病之毒性、胰岛素抵抗的理想模型,通过HE染色后亦可见糖尿病组大鼠心肌的病理形态学改变。
3.2 PI3K-Akt途径与心肌细胞的胰岛素敏感性
PI3K-蛋白激酶B(Akt/PKB)途径介导胰岛素的大部分生物效应,是胰岛素调节细胞生理功能的主要信号通路,可调节葡萄糖摄取、促进细胞分裂、增生及生存、抗凋亡、激活多个基因表达等,PI3K-Akt-mTOR通路是目前研究热点,可调节多种细胞的生理活动,包括细胞的生长、增殖和分化,其过度激活参与糖尿病心肌病、糖尿病肾病、胰岛素抵抗等[12]。本研究发现,正常状态下大鼠心肌组织有PI3K的表达,但2型糖尿病组PI3K的表达较正常组明显减低,提示PI3K的表达与2型糖尿病心肌细胞胰岛素敏感性降低密切相关。在参与基因调控方面,分析可能是由于PI3K的表达减低,导致下游因子Akt的表达变化,由于Akt处于PI3K/Akt通路的中心环节,是PI3K的直接靶基因。许多细胞因子、生长因子和物理刺激等都可通过激活PI3K而使Akt磷酸化,活化的Akt引起下游磷酸化级联反应和靶蛋白之间的相互作用,调控细胞生长与存活、增殖与凋亡,糖类代谢,基因转录等多种细胞活动和生物学效应,而PI3K的表达直接影响下游因子的表达变化。因此,PI3K表达增加可促进外周组织的糖利用,减轻胰岛素抵抗,增强将葡萄糖作为能量来源的能力,使心脏供能加强并防止心功能不全,而PI3K表达下降则影响心肌细胞生理活动。
3.3 PGC-1信号传导途径在心肌代谢中起关键作用
在复杂的转录途径的主要成分中,过氧化物酶体增殖物激活受体γ共激活因子1α(PGC-1α)起关键作用,作为代谢传感器,负责转录的微调对过多刺激的反应[13],并且调节心肌细胞糖酵解到有氧氧化的代谢转换。PGC-1α表达增多明显加强脂肪酸的氧化,并发生在糖酵解的肌肉中[14]。本实验经过高脂喂养并通过STZ诱导的糖尿病大鼠模型中,PGC-1α表达水平升高。PGC-1α表达加强脂肪酸β-氧化,而利用葡萄糖和脂肪酸供能转变几乎全部通过脂肪酸β-氧化提供能量为糖尿病心肌病发生的重要机制之一。此外,低氧刺激PGC-1α表达,而后PGC-1α与核受体相互作用,增加血管内皮生长因子(vascular endothelial growth factor,VEGF)的分泌[15]。我们推测PGC-1α的表达增加,加强糖尿病心肌细胞由葡萄糖供能转变为脂肪酸氧化供能,可能为不利因素。此外,PGC-1α在决定线粒体含量中起主要作用。线粒体增生失控,导致线粒体数量和体积骤增,同时伴有线粒体生物发生相关基因的上调;或许已有线粒体超微结构破坏和病理心肌的发生[16]。综上所述,我们可以推测PGC-1α在心肌细胞中的表达增加可能引起线粒体的增生,数量与体积剧增。线粒体含量的任何增加都会以肌原纤维体积增大为代价,从而使心肌肥大,进一步发展可导致心脏功能失代偿。
3.4 二甲双胍的心血管保护作用
作为2型糖尿病治疗一线治疗药物,许多循证医学证据表明,二甲双胍除了能够降低血糖,改善胰岛素抵抗之外,还具有心血管保护作用,能够减轻缺氧、缺血再灌注损伤、减少心肌细胞凋亡。保护血管内皮、延缓心肌纤维化、改善心肌肥厚的积极作用。二甲双胍能够通过PI3K-Akt依赖途径诱导Akt磷酸化抑制线粒体通透性转运体(mitochondrial permeability transition pore,mPTP)开放来减轻缺血再灌注损伤[17]。近期有文献报道二甲双胍还可以激活Akt1并使JNK3及c-Jun磷酸化水平降低,达到抑制细胞凋亡改善缺血再灌注损伤。其具体分子机制是经由PI3K-Akt-mTOR途径介导抑制缺血心肌细胞凋亡、促进心肌存活,对大鼠心肌细胞发挥正性肌力作用[18]。从本实验可以看出,二甲双胍组较糖尿病组PI3K表达升高,说明二甲双胍可以通过PI3K-Akt介导的途径对糖尿病心肌细胞发挥作用。此外,本实验中二甲双胍干预后心肌细胞PGC-1α表达较糖尿病组降低,提示二甲双胍可能通过PGC-1α表达的减少,减少脂肪酸β-氧化,加强葡萄糖供能,改善心肌代谢。同样近期有研究结果显示大鼠梗死心肌中PGC-1α的蛋白质表达水平上调,细胞凋亡率上升,二甲双胍干预后,PGC-1α蛋白质表达水平下降,细胞凋亡率提升[19],研究结果与本实验一致。
综上所述,二甲双胍干预组大鼠的血脂相关生化指标及胰岛素抵抗指数均较糖尿病组有明显改善,可改善脂毒性及胰岛素抵抗。可能通过抑制大鼠心肌细胞PGC-1α表达,同时上调大鼠心肌细胞PI3K p85的途径完成,改善心肌能量代谢,增强心肌细胞的胰岛素敏感性。PGC-1α与PI3K-Akt途径之间的调节机制,其下游因子之间的相互作用,仍需进一步研究,进而更深入地研究糖尿病心肌病机制及二甲双胍对2型糖尿病患者心脏保护作用机制。
猜你喜欢
临床肺科杂志(2022年3期)2022-11-26
世界中医药(2022年18期)2022-11-25
中华实用诊断与治疗杂志(2022年1期)2022-08-31
生物化学与生物物理进展(2022年5期)2022-05-23
中西医结合心脑血管病杂志(2022年4期)2022-03-11
中西医结合心脑血管病杂志(2022年2期)2022-02-15
小雪花·成长指南(2021年2期)2021-05-20
初中生世界·九年级(2019年4期)2019-05-05
体育科学(2018年12期)2019-01-04
中国体育科技(2018年6期)2018-12-13
