
5-羟甲基糠醛选择性氧化制备2,5-二甲酰基呋喃研究进展
2021-12-16 01:36熊宇杭陈立芳漆志文
化学反应工程与工艺 2021年3期
熊宇杭,陈立芳,漆志文
华东理工大学化工学院化学工程联合国家重点实验室,上海 200237
日益严峻的环境问题和能源危机迫切需要发展绿色可再生能源新结构,含碳资源的清洁转化和高效利用成为能源科技发展的重大战略需求。生物质资源作为唯一可再生的碳基资源,每年浪费高达100亿~500亿吨,合理开发并充分利用生物质能不仅可带来可观的经济效益,而且是循环经济可持续发展的根本路径。5-羟甲基糠醛(HMF)作为一种重要的生物质平台化合物,由可再生资源——生物质碳水化合物脱水转化而来,通过水解、聚合、卤化、酯化、加氢以及氧化还原等反应催化可转化为燃料、高分子材料、农药和医药中间体等高附加值下游产品,是连接生物质资源与化石资源的重要桥梁[1-2]。以农林生物质资源为原料的清洁生产新工艺是实现经济可持续发展的必经之路,而生物质平台化合物取代石油基大宗化学工业原料具有十分广泛的应用前景,是资源、能源和化学工程等领域最具挑战性的研究课题。
HMF可以制备高品质的液体燃料,经氧化制备代表性的产物如5-羟甲基-2-呋喃甲酸(HMFCA)、2,5-二甲酰基呋喃(DFF)、5-甲酰基-2-呋喃甲酸(FFCA)和2,5-呋喃二甲酸(FDCA)等[2-3]。其中DFF是合成医药、有机导体、荧光剂和大环配体等精细化学品的中间体,也是一种重要的呋喃基聚合物单体,还可用于制备聚频哪醇和聚乙烯等[4-5];产物FDCA的结构及性质与对苯二甲酸(PTA)相似,可作为PTA替代物取代聚对苯二甲酸乙二酯(PET)等大宗型聚酯材料的原料[6-7]。作为研究基础,深入认识HMF氧化机理过程有利于HMF的定向转化和新型工业催化剂的设计开发。本综述将以DFF为HMF氧化反应的目标产物,对HMF选择性氧化制备DFF的主要几类催化剂体系进行系统地阐述和总结,并展望未来可能的研究方向和发展趋势。
1 HMF催化氧化产物分布和反应机理
HMF由一个呋喃环、一个醛基及一个羟基组成,其氧化过程实则是醛基和羟基共同氧化的过程,主要产物有HMFCA,DFF,FFCA和FDCA,另外还生成几种副产物。一般来说,较高的反应温度下容易发生呋喃环的聚合而生成HMF聚合物(胡敏素),C—C键清除氧化则生成马来酸酐(Maleic anhydride),酸性条件下易攻击呋喃环而发生开环反应生成顺丁烯二酸(Maleic acid)、反丁烯二酸(Fumaric acid)或乙酰丙酸(Levulinic acid),深入过渡氧化则生成碳的氧化物(CO和CO2),整个氧化反应产物网络如图1所示[8-10]。
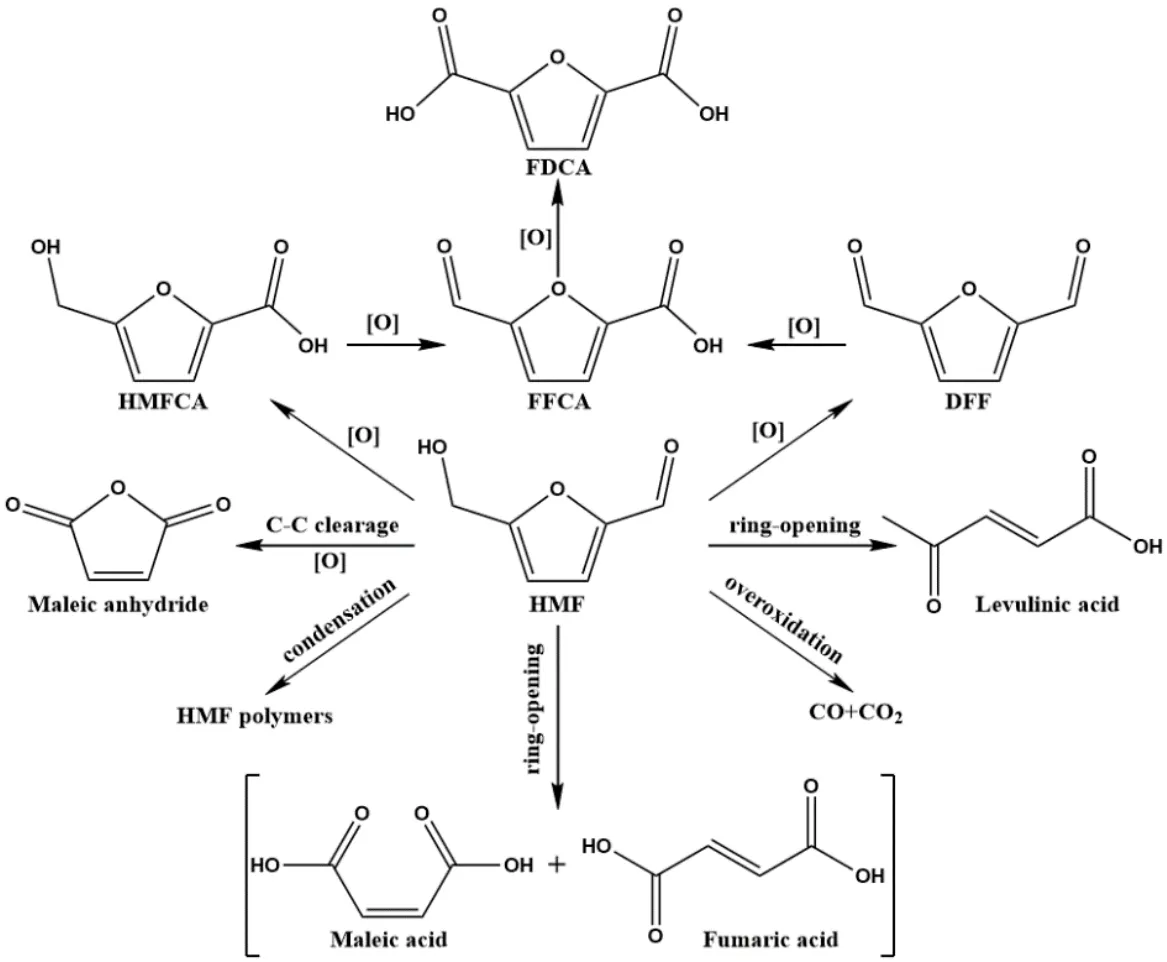
图1 HMF的氧化反应网络[8-10]Fig.1 General reaction network for HMF oxidation[8-10]
在碱性条件下,亲质子性的羰基基团加成、质子转移脱水形成HMFCA,贵金属催化剂则活化HMFCA中—CH2OH基团上的C—H而形成FFCA,再经历羟基加成和脱水形成FDCA[11-13],见图2(a)。非碱性条件下,贵金属催化剂催化HMF氧化的路径是先形成DFF,然后深度氧化为FDCA,主要通过Langmuir-Hinshelwood反应机理,同位素研究显示决速步是贵金属表面氧物种对醇类物中氢的提取[14-15],见图2(b)。正是因为HMF氧化机理方面的深入认识促进了贵金属催化剂的精确调控方法,因而可设计催化性能更优的贵金属催化剂。
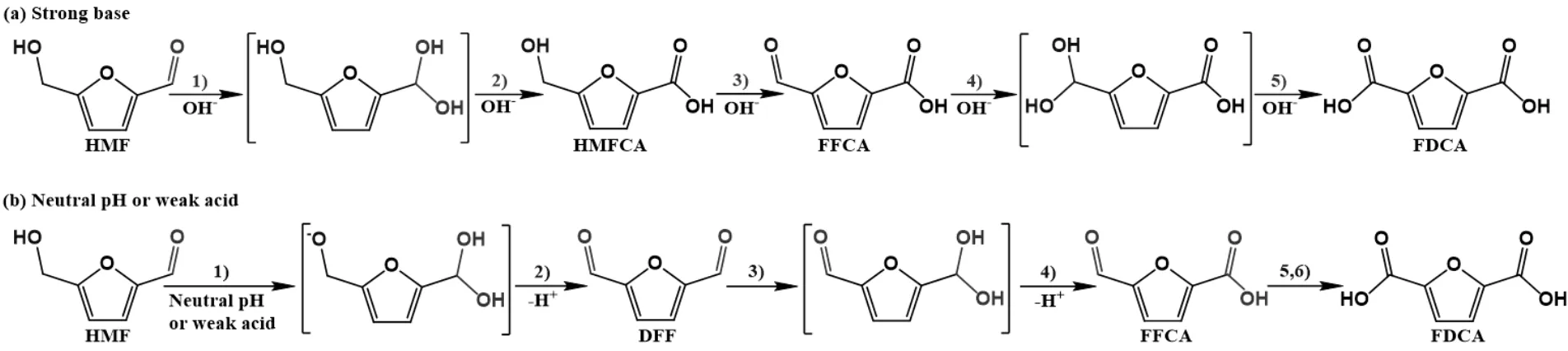
图2 贵金属催化HMF在碱性(a)和弱碱或非碱性(b)介质中的氧化过程[11,14]Fig.2 HMF oxidation based on noble catalysts in strong basic media (a) and slightly basic or neutral media (b) [11,14]
目前催化HMF氧化非贵金属催化剂主要是过渡金属氧化物或者复合氧化物,和贵金属催化HMF氧化类似,在碱性和非碱性条件下的反应路径是不同的[16-19]。若要氧化其中的—CH2OH基团为—CHO基团(目标产物为DFF)时,应避免使用强碱溶液,因为HMF和DFF中的—CHO基团极易氧化成FFCA,再过度氧化为FDCA。非碱溶液的过渡金属催化HMF氧化机理涉及—CH2OH和—CHO基团氧化,晶格氧(Olatt),即和金属配位的氧桥(M-Olatt)参与反应,留下与金属配位的氧空穴(M-#),金属氧化位被还原,通过吸附在金属位分子氧(O2*,*为金属位)分解得到补偿(O2*→2O*),可能的机理如下[19],其中R代表呋喃环。
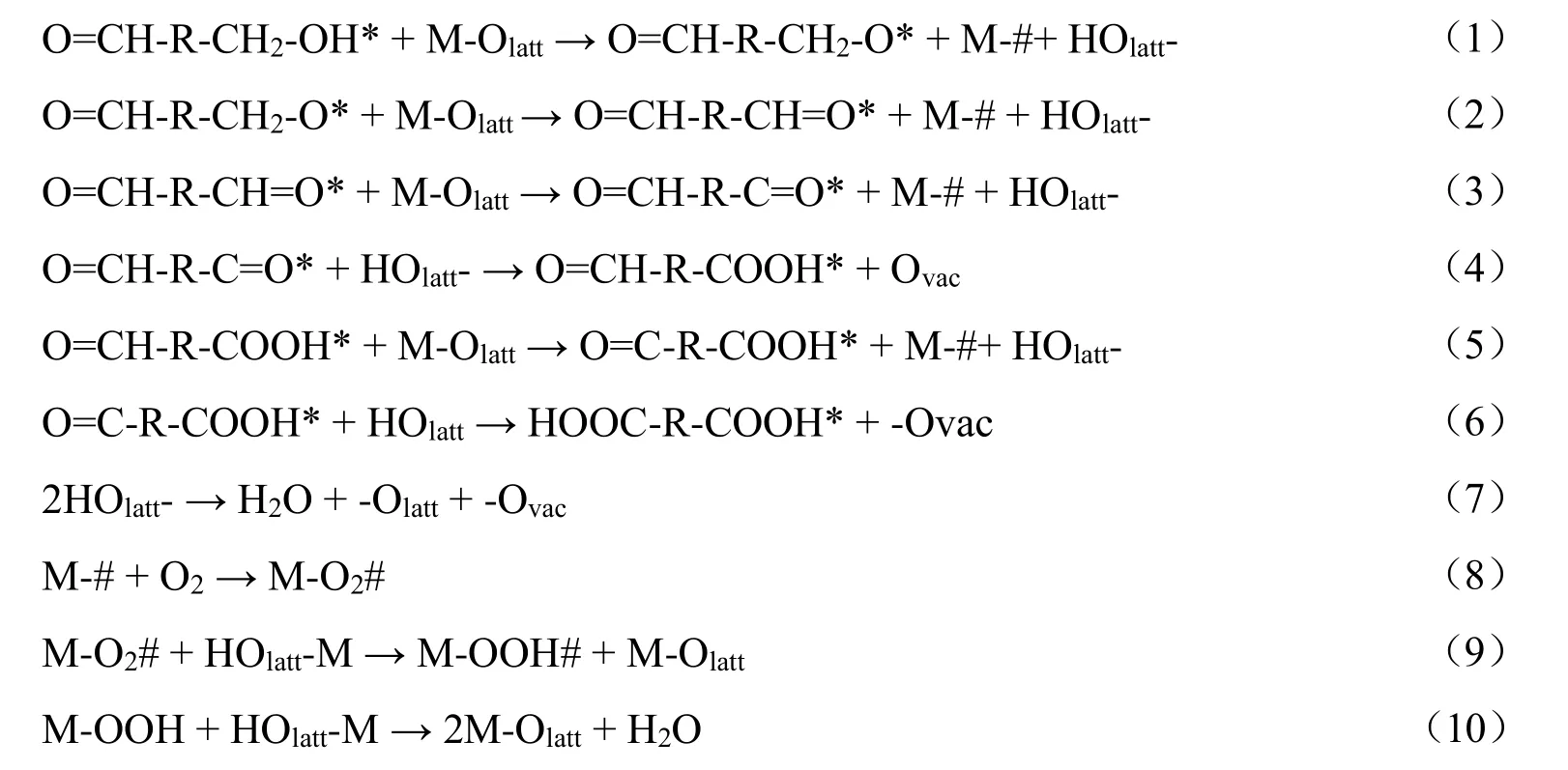
HMF首先和晶格氧作用通过两次氧化脱氢生成DFF,同时产生两个—HOlatt分子,见式(1)和式(2)。DFF中的一个醛基进一步被晶格氧氧化为—CO基团并生成—HOlatt基团,接着—HOlatt基团插入—CO基团形成FFCA,留下氧空穴位(Ovac),见式(3)和式(4)。然后,FFCA上的醛基基团通过类似的基本步骤被氧化为羰基基团而生成FDCA,见式(5)和式(6)。消耗晶格氧生成的—HOlatt通过形成水释放了H并留下一个氧空穴位,见式(7)。或者通过分子氧除去H:氧气首先吸附在金属表面的氧空穴位(M-#),然后和邻近的—HOlatt形成OOH#,生成的OOH#从邻近的—HOlatt中捕获一个氢生成水,消耗晶格氧后留下的氧空穴通过分子氧恢复至晶格氧,且金属氧化物从还原态变为氧化态,完成一次氧化还原循环,见式(8)~式(10)。因此,根据HMF氧化反应路径,优化催化剂结构和组成可获得高产率的目标产物。但是目前的催化技术很难满足严苛的反应条件,本综述的目的是为了促进HMF选择性氧化制备DFF的工业化,对其催化体系提出可能的发展方向和挑战。
2 均相催化体系
早期HMF选择性氧化制备DFF过程主要集中在氧化剂参与反应的传统化学计量反应或均相反应催化体系,如铬酸盐或高锰酸盐。Cottier等[20]用2,2,6,6-四甲基哌啶氮氧化物(TEMPO)作催化剂,对甲苯磺酸(PTSA)作助催化剂催化呋喃醇氧化,并利用水和二氯甲烷两相溶剂的萃取作用提高DFF收率,反应10 min后DFF收率达到81%。McDermott等[21]采用氯铬酸吡啶(PCC)为催化剂和氧化剂,二氯甲烷为溶剂,DFF收率能达到81%。Amarasekara等[22]采用各种有机锰(III)复合物(Mn-Salen)为催化剂,次氯酸钠为氧化剂,在pH值为11.3的磷酸缓冲溶液和二氯甲烷组成的双相体系中进行萃取,室温下反应24 h,DFF收率可达63%~89%。
为了避免使用化学计量氧化剂,H2O2、空气和O2等绿色氧化剂也慢慢被采用。Hansen等[23]利用TEMPO和CuCl的2,2-联吡啶配合物[CuCl(bipy)]为催化剂,乙腈为溶剂,空气为氧化剂,在50 ℃下反应24 h,DFF收率高达90%。Zuo等[10,24]在醋酸中用Co/Mn/Br为均相催化剂,3 MPa的CO2/O2(体积比为1:1)为氧化剂,在180 ℃下反应30 min,主要产物为FDCA,收率为90%。高含量的CO2和酸性反应条件增强了对呋喃环的攻击,导致开环反应而生成乙酰丙酸、马来酸和丁二酸等。研究认为双金属之间氧化还原循环产生的协同作用有利于产生自由基,发生减氢作用,导致DFF过度氧化成FFCA,由于FFCA中羰基基团的吸电子效应使FFCA氧化成FDCA为整个反应的控制步骤。
尽管传统的均相催化体系能实现HMF选择性氧化制备DFF,但反应效果并不理想。另外,该类反应体系具有腐蚀性、产生有毒有害的过渡金属废弃物以及原子利用率低等问题,不符合当前绿色化学发展趋势。因此,寻求高效多相催化反应路线替代均相催化反应、简化分离和资源高效利用是科研人员的研究重点。
3 负载型贵金属催化体系
基于传统氧化剂和均相催化剂的缺陷,选择绿色环保的H2O2、空气和O2等分子氧作氧化剂,在负载型贵金属催化剂上催化HMF选择性氧化。由于贵金属的高活性,在碱性条件下DFF中的醛基基团很容易被深度氧化为FDCA,导致DFF选择性不高,需要通过调变溶剂种类、体系pH值、反应温度、氧气分压和催化剂自身性质等因素来提高DFF选择性。
Nie等[25]比较了活性炭负载的Pt,Rh,Ru,Pd和Au催化剂,发现在以甲苯为溶剂、2.0 MPa氧气和110 ℃反应条件下,Ru/C催化HMF选择性氧化得到DFF最高收率为95.8%。同时还比较了酸碱性位不同的载体如Al2O3,ZSM-5,TiO2,ZrO2,CeO2,MgO和Mg2AlOx,考察了Ru的粒径分布及溶剂体系对HMF氧化产物分布的影响,结果表明载体表面的酸性位和碱性位有利于避免HMF的聚合和降解,当Ru团簇为2 nm时(见图3),Ru/C催化HMF选择性氧化制备DFF活性最好,且经历Langmuir-Hinshelwood机理,HMF和O2形成吸附的醇和原子氧物种,然后吸附的原子氧物种抽提Ru物种表面醇物种上的氢为速控步骤而生成DFF[26]。

图3 的Ru/C催化剂的TEM和粒径分布[26]Fig.3 TEM micrographs and size distributions of Ru/C[26]
Yadav等[27]将Ag+取代纳米纤维结构分子筛中的K+得到Ag-OMS-2。以异丙醇为溶剂,Ag负载量为15%(质量分数)的Ag-OMS-2在1.5 MPa空气氛围,165 ℃下反应4 h,HMF转化率为99%,DFF选择性为99%,且重复使用6次以上没有明显的活性降低现象,该反应的活化能为48.28 kJ/mol。OMS-2中的酸性位负责HMF的副反应,而Ag+的取代不仅有利于降低催化剂表面的酸性位,并且可以快速移除HMF中的第二个氢,从而提高催化性能。
基于HMF氧化路径,可知在碱性条件下贵金属催化剂容易将HMF过度氧化为FDCA。Megías-Sayago等[28]利用离子交换法合成了Au/CexZr1-xO2催化剂,并探讨了在碱性条件下载体表面酸性-碱性位对于生成FDCA的作用机制。结果发现,为了生成高选择性的DFF,需要在无碱条件下催化HMF氧化。Mishra等[29]采用尖晶石型MnCo2O4负载Ru纳米粒子得到了Ru/MnCo2O4催化剂,在1.0 MPa的O2氛围和非碱溶剂甲苯中,130 ℃下反应3 h,DFF产率高达98.3%。Liu等[30]用高能球磨法合成了原子尺度分散的Ru催化剂(Ru/Ni),以甲苯为溶剂,在1.0 MPa的O2氛围和110 ℃下该催化剂催化HMF反应2 h,转化率为91.1%,DFF选择性为81.3%,并且表现出良好的稳定性。
贵金属在非碱条件下催化HMF选择性氧化制备DFF均能达到很好的效果,在温和的反应条件下,HMF转化率和DFF选择性均能达到95%,且重复利用率高。但是在反应条件下催化剂易团聚、活性组分易流失和价格昂贵等缺点限制了其工业化应用。于是,储量丰富、价格低廉的多元过渡金属氧化物催化剂在此反应中显示出诱人的前景。
4 过渡金属氧化物催化剂
过渡金属锰或钒有多变的电子结构,具有高储氧及快速吸附氧的能力,表现出优良的氧化还原性能,近年来广泛用于催化HMF氧化过程。Zhao等[31]将VO2嵌入介孔炭微球中,以二甲基亚砜为溶剂,在常压O2氛围、120 ℃下反应4 h,DFF选择性高达99.9%,活性中心为V4+物种。此外合成了由纳米线组装的钒氧化物微球,催化HMF转化率高达93.7%,DFF选择性为95.4%。实验和理论计算结果均表明钒氧化物暴露(010)晶面的V=O位具有强吸氢能力,有利于HMF分子中的—CH2OH基团活化,生成高选择性的DFF。同位素追踪结果表明,利用V=O位进行αH—C键清除是HMF选择性氧化为DFF的速控步骤。反应机理涉及吸附H的V=O位还原为V—OH及V—OH通过分子氧的再氧化过程,也就是V=O/V—OH循环,见图4[32]。
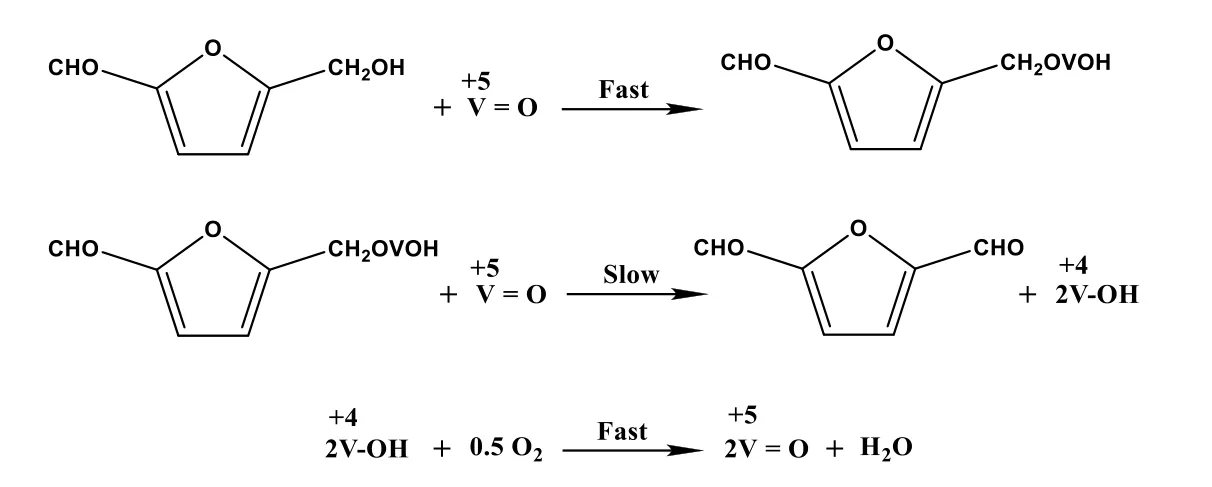
图4 基于VO2+/C催化剂的HMF氧化过程[32]Fig.4 The oxidation of HMF over the VO2+/C catalyst[32]
因为钒氧化物存在一定毒性,则过渡金属锰基氧化物催化HMF选择性氧化制备DFF更具吸引力。Nie等[33]合成了各种形貌的锰分子筛(OMS),其中OMS-2具有2.3 nm×2.3 nm和4.6 nm×4.6 nm隧道结构,以DMF为溶剂,在0.5 MPa O2氛围和110 ℃条件下反应1 h,DFF收率高达97%。该反应为Mn4+/Mn3+氧化还原循环的Mars-van Krevelen机理,同位素追踪结果表明催化剂表面晶格氧吸附的醇物种与邻近CH2之间发生的C—H清除,还原的Mn3+被化学吸附的氧分子再氧化为Mn4+的过程是HMF在OMS-2催化剂表面的动力学相关步骤。Sarmah等[34]利用H-Beta和OMS分子筛结合将二糖或者多糖两步转化为DFF,H-Beta中的Brønsted酸性位有利于糖的水解与脱水,Lewis酸性位有利于异构化得到HMF,OMS分子筛则催化HMF氧化,用甲基异丁酮作萃取剂可获得高收率(97.1%)的DFF。
锰分子筛催化HMF选择性氧化制备DFF具有优良的催化活性,采用单一的锰氧化物催化HMF选择性氧化制备DFF也有大量报道。本研究团队利用微波辅助离子液体路径合成了Mn5O8纳米片,草酸作沉淀剂合成了MnOx微纳米管,两种锰氧化物对HMF选择性氧化制备DFF均表现出有效的催化活性[35-36]。Yu等[37]合成了各种锰氧化物Mn2O3、Mn3O4、无定形MnOx和α-MnO2,催化HMF选择性氧化制备DFF均表现出一定的催化活性。总的来说,单一锰氧化物对HMF选择性氧化DFF的活性和选择性均不高,为了提高其催化活性,复合金属氧化物也广泛应用于HMF氧化过程。
5 复合金属氧化物催化剂
过渡金属锰或钒可分别与钴、铁、镁或铈等形成双金属氧化物催化HMF选择性氧化制备DFF。锰和钒具有多变的电子结构,与另一种金属/金属氧化物在电荷转移过程中使电荷发生变化,导致复合氧化物的几何构型发生畸变产生Jahn-Teller效应,提高了复合氧化物的氧化还原能力,于是提高催化剂的催化性能。
Nie等[38]揭示了钒氧化物负载在载体TiO2,Al2O3,Nb2O5,ZrO2和MgO之后的结构-活性关系。钒氧化物的表面密度和第二种金属氧化物会影响钒氧化物结构和氧化还原性能,从而影响催化活性。第二种金属的加入使得钒氧化物结构变化,引起单层容量内的表面密度增加,导致氧化还原性能提高,而第二种金属的酸性位有利于生成DFF。Ventura等[39]考察了MgO/CeO2混合氧化物结构与HMF选择性氧化制备DFF,FFCA或HMFCA反应性能之间的关系,认为当催化剂表面酸性位和碱性位相当时有利于生成DFF,当HMF浓度较低且催化剂表面碱性位增加则有利于生成HMFCA,而FFCA则随着反应时间的延长而增加。Yang等[40]合成了三维花状Ce-Mo复合氧化物,其扫描电镜(SEM)照片以及相应的元素分布见图5。当Ce/Mo物质的量比为9时,得到的Ce9MoOδ拥有八面体MoO6物种,具有最多的晶格缺陷,展现出最好的催化活性。

图5 Ce9MoOδ复合催化剂的SEM照片(a)和相应的Ce、Mo和O元素分布(b-d)[40]Fig.5 SEM image of Ce9MoOδ (a) and the corresponding EDS element mapping of Ce, Mo, and O (b-d)[40]
对于广泛使用的锰基催化剂,过渡金属铁的加入可增加催化剂表面催化活性位数量,从而提高催化活性[41],当Mn/Fe物质的量比为6时制备的Mn6FeOx,在1.5 MPa O2氛围,110 ℃下催化HMF氧化反应5 h,HMF转化率高达97%,DFF选择性为98%。Yuan等[42]将Cs引入MnOx,不仅提高了催化剂中Mn的氧化态和表面碱性位,还提高了表面的缺陷位并增加了催化剂的比表面积,提高了催化剂的催化活性,在1.0 MPa O2氛围,100 ℃下催化HMF氧化反应12 h,HMF转化率为98.4%,DFF选择性为95%。本研究团队也合成了尖晶石型CoMn2O4空心微球,因为双金属之间的强相互作用能提高催化剂的氧化还原性能和催化剂表面氧空穴,从而提高了催化剂的催化性能[43]。
6 非金属催化体系
关于负载型贵金属和过渡金属氧化物催化剂催化HMF选择性氧化过程已被广泛报道,同时非金属催化体系也得到了很好的发展。2011年,Yoon等[44]报道了聚苯乙烯负载的邻碘苯甲酸铵催化剂,以氯仿为溶剂,在室温下催化HMF氧化反应2 h,HMF转化率为87%,DFF选择性为98%。氧化石墨烯表面缺陷边缘存在的羧酸基团和邻近未成对电子协同作用可增强分子氧的捕获和活化,导致TEMPO催化剂的电子转移至分子氧,有利于HMF选择性氧化制备DFF。于是Lv等[45]采用氧化石墨烯和TEMPO共催化体系,在常压O2氛围和100 ℃下催化HMF反应9 h,发现HMF可完全转化且全部转化为DFF。
非金属催化剂表面不能发生氧化还原反应,因而需要共催化剂TEMPO或其它能与氧作用的催化剂共同作用才能催化HMF氧化[44-45]。Ren等[46]用硝酸促进非金属催化剂的还原与再氧化过程,合成了氮惨杂的碳材料(NC)用于催化HMF氧化。反应过程中碳材料中惨杂的NO3-物种可以氧化HMF,自身则被还原为NO2物种,通过与碳材料表面吸附的分子氧作用再氧化为NO3-,也就是NO3-物种作为活性位与NO2物种通过氧化还原循环而催化HMF选择氧化制备DFF,见图6。该催化剂在1.0 MPa O2氛围和100 ℃下催化HMF反应14 h,DFF的产率高达95%,并且表现出优良的稳定性能。
7 光催化体系
非贵金属用于催化HMF选择性氧化制备DFF报道不多,仍在发展阶段。石墨相氮化碳不仅可直接催化HMF氧化,而且是一种典型的聚合物半导体材料,其结构中CN原子以sp2杂化形成高度离域的π共轭体系,禁带宽度约为2.7 eV,可吸收太阳光中的部分光源,于是清洁、经济的可见光光催化HMF氧化过程近年来也逐渐引起了关注。Wu等[47]报道了用石墨相氮化碳作光催化剂,在氧气流速为10 mL/min,300 W氙灯作光源下催化HMF氧化。在半导体材料边缘光生电子将分子氧还原成·O2-作为活性物使催化剂表面HMF的醇基基团去质子化,生成的烷氧基阴离子(CHO—R—CH2O—)与空穴反应,通过电子转移形成烷氧自由基(CHO—R—CH2O·),该自由基再与过氧化氢自由基结合释放H2O2并生成产物DFF。Krivtsov等[48]合成了比表面积较大(169 m2/g)的石墨相氮化碳光催化剂,以水作溶剂,直接利用太阳光作光源催化HMF氧化,当HMF转化率为40%时,DFF选择性为50%。
自从发现石墨相氮化碳可作光催化剂催化HMF氧化后,各种半导体金属氧化物也渐渐被发现可作光催化剂催化HMF氧化反应过程。Zhang等[49]直接将铌酸焙烧得到Nb2O5,发现吸附在Nb2O5表面的醇羟基基团在可见光照射下一个电子被转移至导带上,Nb5+被还原为Nb4+,同时在醇羟基上留下一个光生空穴,形成的烯基自由基转化为羰基化合物DFF从催化剂表面脱附,而还原的Nb4+位通过分子氧再氧化为Nb5+,继续进行下一次氧化还原循环。Zhang等[50]合成了钙钛矿型溴化铅甲铵(MAPbBr3),以乙腈为溶剂,在室温和常压空气氛围下,用LED蓝光照射催化HMF氧化,结果表明HMF可完全转化,且DFF选择性高达90%。Khan等[51]利用可见光原位光敏化二氧化钛催化剂,可见光催化HMF氧化反应4 h,HMF转化率为50%,DFF选择性为87%。Dhingra等[52]合成了Z型的光催化体系(Zn0.5Cd0.5S/1%MnO2),利用30 W的可见光LED灯作光源,水作溶剂,室温下光催化HMF氧化制备DFF并耦合产氢,反应24 h后DFF产率为46%,选择性为100%,H2产生量为1.322mmol/g,见图7。

图7 Zn0.5Cd0.5S/1%MnO2光催化HMF氧化并耦合产氢过程[52]Fig.7 Process of HMF oxidation to DFF coupled H2 production by visible-light-driven over Z-scheme Zn0.5Cd0.5S/1%MnO2[52]
8 电化学氧化体系
电化学催化HMF氧化合成DFF是另一种逐渐发展的新方法。Kokoh等[53]在碱溶液中利用Pt/Pb电催化HMF氧化,DFF收率为80%。Kashparova等[54]利用双铂电极将HMF溶于含有KI的碳酸氢钠缓冲溶液和乙酰氨基-TEMPO的二氯甲烷溶液组成的双相萃取体系进行HMF电化学氧化,DFF收率约为60%。Kisszekelyi等[55]用石墨棒做阳极,不锈钢做阴极,在TEMPO和二甲基吡啶协同作用的乙腈电解质溶液中催化HMF电化学氧化,DFF选择性高达100%,收率约为80%。二甲基吡啶通过氢键极化HMF中的醇羟基使反应活化能降低58%,于是氢质子从HMF转移至催化剂TEMPO过渡态后发生完整的氢转移生成DFF,见图8。
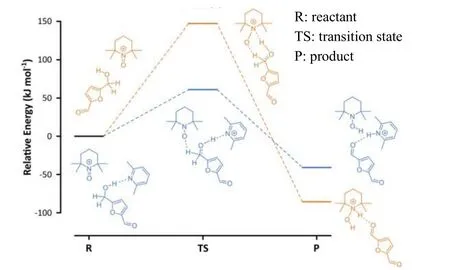
图8 TEMPO和二甲基吡啶协同作用的HMF电氧化能谱[55]Fig.8 Computational energy spectrum of HMF electrooxidation by the synergistic effect of TEMPO and 2,6-lutidine[55]
上述电化学氧化反应需要使用TEMPO作引发剂,通过改进电极材料可避免使用严苛的电解质溶液,是环境友好的电化学过程。Cao等[56]合成了平均粒径为3~6 nm的Pt,Pt-Ru和Ru颗粒负载在碳布上作阳极,0.5 mol/L硫酸溶液作电解质,在50 ℃下HMF电化学氧化反应17 h,DFF选择性为89%。Zhang等[57]利用共沉淀法合成了层状的复合NiCoFe插层结构氢氧化物(NiCoFe-LDHs)采用NiCoFe-LDHs作工作电极,Pt丝为对电极,Ag/AgCl为参比电极的三电极法,在含有饱和氧气的1 mol/L NaOH电解质溶液中,55 ℃下HMF电化学氧化反应1 h,HMF转化率高达95%。因为电解质溶液呈碱性,生成的主要产物为FDCA,结果符合HMF在碱性条件下的氧化反应过程。
9 结论与展望
HMF作为生物质资源与化石能源的桥梁,可催化转化为高附加值化学品和液体燃料,是一种具有应用前景的生物质平台化合物。对于HMF选择性氧化制备DFF过程,均相催化体系由于高能耗的分离和提纯限制了HMF大规模的工业生产,多相催化体系易分离、可循环利用的优势被广泛应用,但仍存在活性组分流失的问题。HMF的高选择性氧化是现代生物炼制技术的核心,高活性的催化剂和经济有效的分离提纯是DFF工业化生产的关键。
为了发展绿色化工过程,未来研究中还需要进一步优化相关催化体系,设计更加绿色高效的多相催化体系,开发新的功能性绿色溶剂和高效分离技术,研究溶剂与催化剂的作用规律,实现溶剂与催化体系的高效分离。其它具有应用潜能的催化技术,如光催化、电化学及微波合成等高效、绿色和可持续发展的合成方法仍处于探索阶段。开发HMF选择性氧化制备DFF及耦合产氢技术,构建完整的从生物质到高附加值化学品和燃料的工艺流程,以实现生物质原料的有效利用。
猜你喜欢
世界农药(2022年10期)2022-11-10
炼油技术与工程(2022年8期)2022-08-18
能源化工(2021年2期)2021-12-30
大连民族大学学报(2021年5期)2021-11-15
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
农药科学与管理(2021年2期)2021-03-16
陶瓷学报(2020年6期)2021-01-26
新课程·下旬(2019年7期)2019-09-17
发明与创新·中学生(2017年11期)2017-12-07
