
唑烷酮类抗耐药菌新药
——康替唑胺
2021-12-15 01:15:56王星海
中国感染与化疗杂志 2021年6期
袁 红, 王星海, 张 菁
多重耐药革兰阳性菌是致细菌性感染的重要病原菌之一,其感染具有高发病率和高病死率。临床最为关注的革兰阳性病原菌为耐甲氧西林金黄色葡萄球菌(MRSA),耐甲氧西林凝固酶阴性葡萄球菌(MRCNS)和耐万古霉素肠球菌(VRE)。当前治疗上述耐药菌感染的抗菌药物,包括糖肽类、唑烷酮类等多种药物已用于临床,然而既安全又有效的抗菌药物仍未能满足临床需求。
1 康替唑胺的设计与筛选
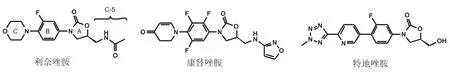
图1 三种唑烷酮类化合物的化学结构
表1 三种唑烷酮类化合物体外抗菌活性和安全性对比(mg/L)

表1 三种唑烷酮类化合物体外抗菌活性和安全性对比(mg/L)
抗菌药物MIC90 (MRSA)CD34+ 抑制IC50 MAO-A 抑制IC50利奈唑胺 1 6.3~7.9 4.1~4.9特地唑胺 0.5 0.2 1.8康替唑胺 0.5 15.7 12.3
2 作用机制
细菌蛋白质合成包括起始、延长及终止阶段,其中,起始阶段需由70S亚基/30S亚基、mRNA及起始型甲酰蛋氨酸tRNA(tRNAfMet)形成的复合物参与。不同于传统的肽酰转移酶抑制剂,唑烷酮类与50S亚基结合,从而抑制起始复合物的形成,而不直接阻碍tRNAfMet的形成[7],这一特殊的作用机制,使唑烷酮类不会与其他抗菌药物发生交叉耐药现象[5]。
从利奈唑胺与金黄色葡萄球菌核糖体结合的单晶结构可以看出[10],利奈唑胺结构中苯环(B环)和唑烷酮环(A环)接近于平面结构。而康替唑胺结构与利奈唑胺的主要区别,在于B环A环的邻位引入了一个氟原子,氟原子具有比氢原子更大的体积和吸电子性,导致康替唑胺B环-A环的夹角要明显大于利奈唑胺,使得分子在空间上具有更大的折角。该A-B环非平面结构被认为能够使康替唑胺避免传统唑烷酮类化合物的联苯平面结构带来的毒副作用[5]。从康替唑胺的构效关系研究中也发现,邻位氟原子的引入,使其对人体单胺氧化酶的抑制作用降为原结构的5%以下[5]。
3 药效学
3.1 体外抗菌作用
康替唑胺的体外抗菌活性研究显示,该药对革兰阳性菌包括葡萄球菌属、链球菌属、肠球菌属以及棒状杆菌属等细菌均显示了高度的抗菌活性,包括耐药菌株如甲氧西林耐药的葡萄球菌(MRSA、MRSE)、青霉素不敏感肺炎链球菌(PISP和PRSP)和VRE[11-12]。见表2。
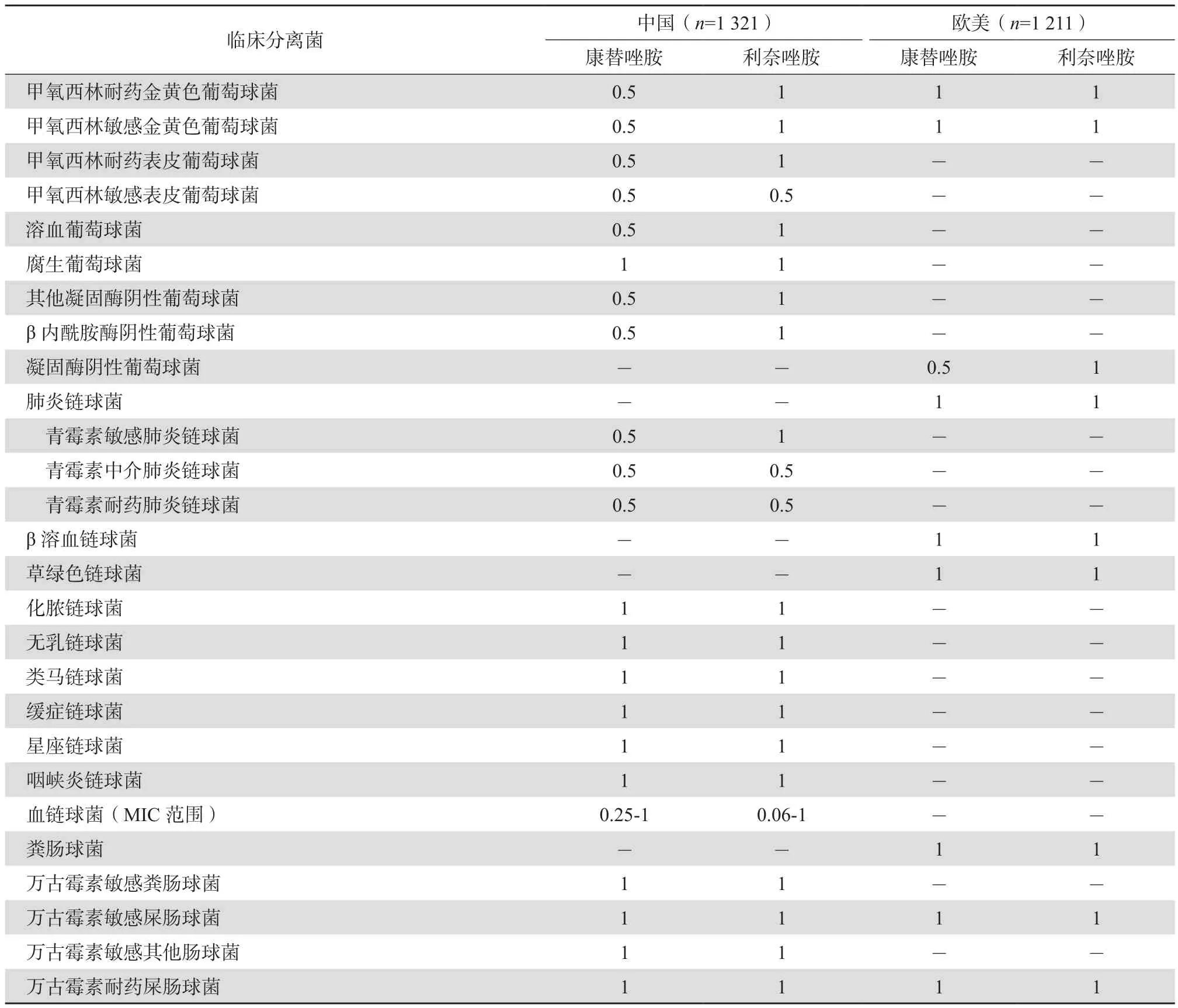
表2 康替唑胺体外抗革兰阳性菌的MIC90值比较(mg/L)
康替唑胺对中国1 321株革兰阳性临床分离菌的体外药效学研究显示[11],与其他受试药相比,康替唑胺对葡萄球菌属细菌,包括MRSA的抗菌活性相仿或略高于利奈唑胺、万古霉素和替考拉宁。对链球菌属细菌的抗菌活性与利奈唑胺相仿,略低于万古霉素和替考拉宁;对肠球菌属细菌的作用亦与利奈唑胺相仿。康替唑胺对万古霉素耐药屎肠球菌的作用与利奈唑胺相仿,明显高于万古霉素和替考拉宁。康替唑胺对肺炎链球菌有明显的杀菌作用,杀菌活性与利奈唑胺相似,对受试的大部分葡萄球菌有一定的杀菌作用,但对肠球菌属仅具抑菌作用。
康替唑胺对美国和欧洲1 211株革兰阳性临床分离菌的体外药效学研究显示[12],康替唑胺是治疗金黄色葡萄球菌、凝固酶阴性葡萄球菌、肠球菌和链球菌 (包括MRSA和VRE)感染的有效选择。此外,康替唑胺可能对某些重要临床分离菌的抗菌活性略高于利奈唑胺。
3.2 抗生素后效应(PAE)
康替唑胺对金黄色葡萄球菌和肺炎链球菌各3株临床分离菌的PAE测定结果显示[11],均有较明显的PAE。当康替唑胺药物浓度为2倍MIC时,对肺炎链球菌PAE的均值为(0.87±0.24)h,当浓度升至4倍和8倍MIC时,康替唑胺对肺炎链球菌的PAE均值分别延长至(1.60±0.49)h 和(2.84±0.82)h。同样,当康替唑胺浓度为2 倍MIC时,对金黄色葡萄球菌的PAE为(1.23±0.25)h,当浓度升至4 倍和8 倍MIC 时PAE 的均值分别延长至(1.68±0.28)h 和(1.78±0.52)h。康替唑胺对肺炎链球菌和金黄色葡萄球菌均与利奈唑胺一样具有良好的PAE,且随着药物浓度的升高,PAE 延长。
3.3 耐药趋势
康替唑胺不容易诱发耐药,天然耐药频率很低(<8.25×10-12),与利奈唑胺相当[13]。在一项对金黄色葡萄球菌ATCC 29213连续传代试验中,经过30代连续诱导,如图2所示,利奈唑胺的MIC从2 mg/L增加至256 mg/L(128倍),而康替唑胺的MIC仅仅增加至16 mg/L(8倍)。因此,金黄色葡萄球菌对康替唑胺可能具有更低的耐药趋势[13]。
为计算特征值及累积方差贡献率,运用SPSS22.0中因子提取的主成分方法求解因子载荷矩阵。一般情况下累积贡献率达到不低于80%的水平效果最为显著。结果如表2显示,有两个因子的特征值均大于1,除此之外的特征值都接近于0,且因子1与因子2的方差贡献率累加已达到88.6%的较高水平,这说明前两个因子就可以传递原数据88.6%的信息量,使用其进行分析能够得到良好的计算结果。

图2 康替唑胺作用下金黄色葡萄球菌ATCC 29213连续传代的耐药趋势
3.4 体内药效
在小鼠腹腔感染模型中[11,14],灌胃给予康替唑胺后,对各种革兰阳性菌包括耐多药菌株所致的小鼠全身感染模型均显示了良好的疗效,如康替唑胺对金黄色葡萄球菌MRSA(ATCC 33591)、MRSA/VISA(Mu50) 的 ED50分 别 为2.36、2.50 mg/kg,显著低于利奈唑胺的ED507.55、5.35 mg/kg,差异具统计学意义(P<0.01);对MSSA(ATCC 29213)、MSSA(15) 和 MRSA(0850) 的 ED50分 别 为 7.98、5.74 和 3.71 mg/kg, 与 利 奈 唑 胺 的 9.03、5.86、3.63 mg/kg 相似(P>0.05)。对肺炎链球菌,包括PSSP、PISP(ATCC 49619)和PRSP菌株,以及粪肠球菌VSE (ATCC 29212)、VRE (EFL4041)和HLGRVSE HH22(HH22)的小鼠全身感染模型的体内抗菌活性测定实验亦显示,康替唑胺有较好的疗效。小鼠腿部金黄色葡萄球菌感染后在2 h和6 h分别经灌胃给予康替唑胺和利奈唑胺。如图3所示,与空白对照组相比,康替唑胺各剂量组(n=10)腿部组织金黄色葡萄球菌的菌数对数值均显著低于空白对照组(P<0.01)。结果显示,经灌胃给予康替唑胺对小鼠腿部感染金黄色葡萄球菌均有较好的体内抗菌活性,在低剂量组(5 mg/kg)康替唑胺对金黄色葡萄球菌的活性略优于利奈唑胺(P<0.01),其他剂量组疗效同利奈唑胺。以利奈唑胺为对照的康替唑胺体内药效学研究显示,在全身和局部感染模型中,康替唑胺均显示出与利奈唑胺相同或略优的抗菌效果[14]。

图3 康替唑胺对金黄色葡萄球菌腿部感染模型的抗菌效果
4 动物PK
动物PK研究结果显示[15],康替唑胺吸收迅速,达峰时间短,Tmax平均约为0.5~2.6 h。康替唑胺经口重复给药7 d后,在大鼠和犬体内均未见显著的蓄积趋势。
康替唑胺灌胃给予大鼠后,原形药物主要分布在消化道、肺和肾等,药物相关物质主要分布在消化道、肝、肾、肺和肾上腺等组织,在眼球中分布很少。大鼠皮下微透析研究结果显示,康替唑胺单次灌胃给予大鼠,吸收后可快速分布到皮下组织,皮下组织和血浆中游离药物的暴露量均随着剂量的增加而增大,在15 mg/kg剂量下,皮下组织中游离康替唑胺的暴露量与血浆中游离康替唑胺的暴露量相当(相关信息来源于尚未发表的康替唑胺申报注册上市阶段的申请资料)。
5 毒理学
在临床前研究中,康替唑胺显示了良好的安全性[17]。在大鼠急性毒性实验中,未观察到有害作用的剂量达到了5 000 mg·kg-1·d-1。在以利奈唑胺为阳性对照,大鼠灌胃给予康替唑胺4周及恢复期4周试验中,因为无明显毒性,康替唑胺高剂量组由第一、二周的200 mg·kg-1·d-1在第三、四周升至300 mg·kg-1·d-1。康替唑胺组各剂量组未见死亡,利奈唑胺组200 mg·kg-1·d-1剂量下雌鼠均死亡,且在100 mg·kg-1·d-1剂量下给药期末出现明显体重下降、摄食量下降和血小板计数下降[5,17],康替唑胺组 100 mg·kg-1·d-1剂量下体重摄食量及血小板均无变化。康替唑胺和利奈唑胺灌胃给予大鼠的未见有害作用的剂量(NOAEL)分别为 100 和 20 mg·kg-1·d-1。
在另一项大鼠连续13周灌胃给予康替唑胺的长毒性试验中,在 50、100、150 mg·kg-1·d-1剂量下未见与供试品相关的动物死亡,且无眼科、脏器重量、解剖和组织病理学的变化,康替唑胺的NOAEL 为 100 mg·kg-1·d-1。证明大鼠给药周期从4周延长到13周,康替唑胺没有显著增加不良反应的风险(相关信息来源于尚未发表的康替唑胺申报注册上市阶段的申请资料)。
6 I期临床试验
6.1 中国Ⅰ期临床试验
康替唑胺片中国Ⅰ期临床研究共入组112例健康中国受试者,男女各半[18]。见表3。第一部分为随机、双盲、安慰剂对照的单剂递增空腹口服康替唑胺片(50~1 800 mg,共8个剂量组, 其 中 200 mg、400 mg、800 mg、1 200 mg剂量组采集血尿样进行初步PK)研究,入组56例受试者。研究结果显示,单剂递增的耐受剂量范围为50~1 800 mg,最大耐受剂量为1 800 mg。在该剂量范围内不良反应少见而轻微,并呈一过性,受试者耐受性和安全性良好。受试者空腹口服康替唑胺,在200~1 200 mg剂量范围内呈非线性PK特征。
表3 中国健康受试者单剂口服及多剂进餐口服康替唑胺片后的平均PK参数 (SD)

表3 中国健康受试者单剂口服及多剂进餐口服康替唑胺片后的平均PK参数 (SD)
*多剂给药的PK参数由给药第15日的0~48 h数据计算。
参数 单剂给药(空腹) 食物影响 多剂进餐给药*300 mg600 mg900 mg900 mg (空腹)900 mg (高脂餐)600 mg (普通餐)800 mg (普通餐)Cmax / (mg/L)8.07(3.17)12.24(3.59)15.25(3.18)18.10(4.52)42.25(7.11)20.61(5.80)26.45(6.73)Tmax /h 1.54(0.81) 2.02(0.69) 1.61(0.85) 1.44(0.48) 1.63(0.52) 0.60(0.17) 0.57(0.12)AUC0-t / (mg·h/L)29.13(8.52)48.16(13.50)59.16(12.23)66.18(16.79)141.14(17.59)72.91(33.46)90.38(34.26)AUC0-inf / (mg·h/L)29.21(8.54)48.27(13.47)59.60(12.74)66.57(16.83)141.33(17.53)73.61(33.69)92.23(34.62)T1/2 /h 2.08(0.87) 3.40(1.75) 4.25(1.95) 4.50(2.97) 2.10(0.99) 4.02(1.09) 4.84(1.74)MRT0-inf /h 3.57(0.78) 4.88(1.61) 4.72(0.69) 4.07(1.62) 3.21(0.54) 4.22(0.95) 4.87(1.02)CLZ/F/ (L·h-1·kg-1)0.18(0.05)0.21(0.05)0.25(0.06)0.25(0.09)0.11(0.02)0.15(0.04)0.17(0.04)
第三部分为随机、双盲、安慰剂对照的多剂进餐口服康替唑胺(600 mg和800 mg, 第1日及第15日为每日1次服药,第2~14日为1次/12 h口服药1次,连续15 d)的耐受性试验及PK研究,入组32例受试者。研究结果显示,除600 mg组1例受试者的ALT、AST升高程度属中度不良事件外,其他所有与研究药物相关的临床不良事件和实验室检查异常均为轻度。所有不良事件均为一过性,未经处理均自行恢复正常。Cmax和AUC0-24h随剂量等比增加,在人体内皆无蓄积。
口服康替唑胺单剂及多剂耐受性试验结果显示,与研究药物相关的临床不良事件和实验室检查异常均少见而轻微,并呈一过性,受试者耐受性良好。与研究药物相关的临床不良事件主要为恶心、呕吐等消化道症状,与研究药物相关的实验室检查异常,主要为肝酶升高。上述耐受性试验显示,不同于其他唑烷酮类抗菌药物,康替唑胺在单剂和多剂耐受性试验中对血液系统均无明显影响。
PK/PD 分析显示,基于蒙特卡罗模拟,口服康替唑胺的两种给药方案:600 mg 和800 mg,2次/d,14 d(与食物同服),对复杂性皮肤及软组织感染(cSSSI)主要革兰阳性病原菌包括耐多药金黄色葡萄球菌、化脓性链球菌等均可达近100%药效学靶值,预期此两种给药方案用于cSSSI均可获良好的临床和微生物学疗效。
根据康替唑胺片Ⅰ期临床试验研究结果,并参考该药的PK/PD指数分析,建议Ⅱ期临床试验的适应证为cSSSI,剂量探索可分为两个剂量组,600 mg 2次 /d或 800 mg 2次 /d,疗程 7~14 d,给药时宜与食物同服。
6.2 澳大利亚Ⅰ期临床试验
在澳大利亚开展了以利奈唑胺为对照在健康受试者中的随机双盲Ⅰ期临床试验[19],共入组66例受试者。见表4。第一部分为随机、双盲、安慰剂对照的单剂递增设计,入组30例受试者,剂量分别为400 mg、800 mg和 1 200 mg。研究结果显示,安全性数据未见与剂量相关的趋势,进餐状态下不良事件发生率高于空腹状态。
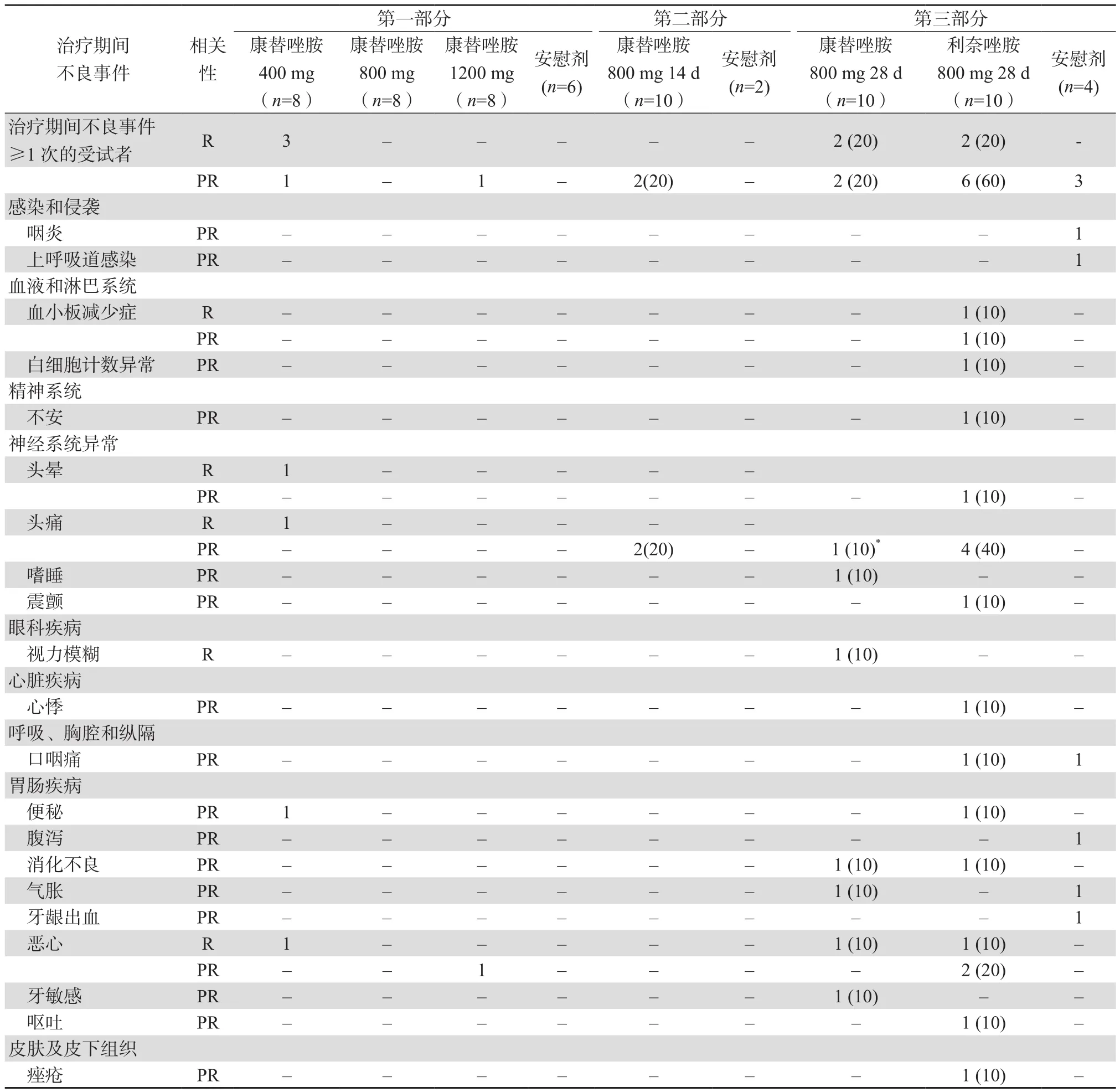
表4 澳大利亚健康受试者口服康替唑胺后与研究药物相关的治疗期间不良事件[n(%)]
第二部分为随机、双盲、安慰剂对照的多剂给药设计,入组12例受试者,受试者口服800 mg康替唑胺片或安慰剂,2次/d,持续14 d。由于受康替唑胺单一剂量限制,未做相关安全性分析。
第三部分为随机、双盲、阳性对照、双模拟的多剂给药设计,入组24例受试者,受试者口服800 mg 康替唑胺片、600 mg利奈唑胺片或安慰剂,
2次/d,持续给药28 d。与研究药物可能相关的不良事件,康替唑胺组4例、利奈唑胺组8例和安慰剂组3例。与康替唑胺相关的不良事件包括轻度的胃肠道不适和中度的头痛;与利奈唑胺相关的不良事件包括轻度的头痛、咽痛和消化不良,中度头痛以及震颤、心悸、恶心、呕吐、血小板下降和白细胞下降。实验室检查显示,康替唑胺800 mg 2次/d连续口服28 d未见明显的血小板计数下降,而利奈唑胺600 mg 2次/d组有5例(50%)受试者血小板计数较基线显著下降(≥1级)。
康替唑胺组受试者均未发生与血液学相关的不良事件或停药,然而, 2例利奈唑胺组受试者发生了3例次血液学相关的不良事件( 2例次血小板减少,1例次白细胞计数减少),其中1例提前终止使用利奈唑胺。另外,相比康替唑胺组,利奈唑胺组的血红蛋白、中性粒细胞和血小板等血液学参数从基线降至最低值≥1级的不良事件发生率较高[19]。
受试者无论是空腹或进餐、单剂或多剂口服康替唑胺后,尿液中原形药物康替唑胺的回收率都很低(≤1%);受试者多剂口服14 d或28 d康替唑胺后,未见康替唑胺及其主要代谢产物M2有明显蓄积。连续口服康替唑胺 28 d的安全性和PK数据表明,康替唑胺有望成为治疗耐药革兰阳性菌所致感染的药物之一。
6.3 TQT研究
康替唑胺全程QT(TQT)研究采用单中心、莫西沙星为阳性对照,安慰剂对照的设盲(莫西沙星组除外)、随机、单中心、4周期、4交叉设计(简称“4交叉设计”),入组52例健康中国受试者[20]。QTcF时间匹配分析是ICH E14推荐的主要终点,QTcF的时间点分析结果显示[20],莫西沙星组在给药后的第3和第4小时的置信区间下限≥5 ms,符合E14检测灵敏度标准,QTcF间期各个时间点安慰剂调整的均值与基线值变化的曲线见图4。研究结果显示,受试者进餐口服1 600 mg康替唑胺,耐受性良好,不良事件多为轻度,所有不良事件均自行恢复。健康受试者口服康替唑胺800 mg(治疗剂量)不会延长经安慰剂校正的QTc间期。

图4 各个时间点安慰剂和基线校正的QTcF间期(ΔΔQTcF) 间期变化
7 结语
康替唑胺是我国自主研发、具有自主知识产权、全新分子实体的唑烷酮类抗菌新药,与已上市的同类药物利奈唑胺具有相似的分子结构和作用机制。临床前研究显示,康替唑胺具有良好的抗菌活性,产生耐药的趋势可能低于利奈唑胺。在国内外进行的Ⅰ期临床试验中,康替唑胺人体耐受性和安全性良好。相比利奈唑胺,Ⅰ期临床研究提示康替唑胺引起血液学毒性的风险更低,有望通过进一步的临床评价,为临床提供可口服且更加安全的抗菌药物新选择。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30 06:39:48
特别健康·下半月(2019年6期)2019-08-01 01:45:35
祝您健康(2019年3期)2019-03-22 08:57:08
兽医导刊(2019年1期)2019-02-21 01:14:06
时代英语·高一(2018年5期)2018-11-19 10:55:06
创新作文(1-2年级)(2018年2期)2018-09-13 11:45:46
创新作文(小学版)(2018年4期)2018-07-06 08:16:00
时代英语·高一(2017年5期)2017-11-14 15:52:20
学习报·教育研究(2017年33期)2017-08-31 02:59:41
兽医导刊(2015年9期)2016-01-04 12:00:06
