
症状前期进行性肌营养不良患儿24例病例系列报告
2021-12-14 09:01曾兴颖吴华平虞雄鹰刘志强
中国循证儿科杂志 2021年5期
曾兴颖 吴华平 曾 苗 虞雄鹰 刘志强
进行性肌营养不良(PMD)是一组原发性肌肉疾病,临床主要表现为进行性肌力减退和肌肉萎缩,常见于男性,主要包括Duchenne型肌营养不良(DMD)和Becker型肌营养不良(BMD)。DMD和BMD均由编码抗肌萎缩蛋白(dystrophin)的DMD基因突变引起。DMD基因突变,若不改变开放阅读框,则能形成缩短的dystrophin蛋白,临床表现为较温和的表型(BMD);若改变了转录本的翻译阅读框或产生终止密码子,则不能形成功能性dystrophin蛋白,临床出现严重的表型(DMD)[1,2,3]。DMD发病率为1/5 000,约是BMD的3倍,属于最常见的儿童神经肌肉性疾病[1],目前缺乏有效的治愈方法。既往临床上常根据患儿丧失行走能力的时间是否早于16岁区分DMD和BMD[1],两者在临床表现、寿命、长程管理和激素的治疗剂量方面均有所不同,早期识别和区分DMD和BMD具有重要的临床价值。本文对江西省儿童医院(我院)收治的婴幼儿PMD病例进行回顾性分析,总结PMD患儿症状前期的特点,旨在早期识别DMD和BMD,以提供更好的疾病管理措施。
1 方法
1.1 伦理和知情同意 本研究获得我院医学伦理委员会批准(伦理审批号:JXSETYY-YXKY-20190035)和患儿监护人的书面知情同意。
1.2 诊断标准 根据文献报道和多个在线网站对DMD基因突变的描述[1,4],结合生化评估[5],本文采用以下婴幼儿PMD诊断标准:①血清CK在病程早期甚至生后即增高,达正常值的10~50倍;②肌电图提示肌源性损害;①和②为临床诊断;③DMD基因变异位点经ACMG评分评为致病性变异为确诊指标。
1.3 研究对象的纳入标准 ①2016年1月至2020年7月在我院神经内科住院或门诊就诊、符合PMD诊断的连续患儿;②年龄>28 d且≤3岁;③完善DMD基因检测。
1.4 基因检测
1.4.1 DMD基因检测方法 经监护人知情同意后抽取患儿外周静脉血2~3 mL,采用多重连接探针扩增(MLPA)技术筛查DMD基因 79个外显子缺失和(或)重复突变。如果样本未检测到外显子缺失和(或)重复突变,通过二代测序法对DMD基因启动子区、5’非编码区(5’UTR)、编码区、转录终止区、内含子剪切点附近10 bp以及有文献报道或数据库记录的已知突变序列进行测序。
1.4.2 基因诊断标准[1]根据阅读框规则,框移突变导致DMD,而框内突变导致BMD,参考网址http://edystrophin.genouest.org/对突变位点进行文献检索结合Dystrophin蛋白的功能预测进行最终诊断。
1.5 分组 根据诊断年龄分为婴儿组(>28 d且≤1岁)和幼儿组(~3岁)。根据基因检测结果,分为DMD组(DMD基因移码突变、无义突变、经典剪切位点突变)和BMD组(DMD基因整码突变、错义突变),若有文献或网站预测的特殊情况,则另行记录。
1.6 临床资料提取 设计统一的临床资料收集表,查阅我院电子病历系统,采集:①患儿的年龄、发育情况、四肢肌力、Gower征、家族史、入院主诉、诊疗经过;②基因检测结果;③血清酶水平:CK-MB、CK、LDH、ALT和AST;④肌电图。
1.7 统计学方法 采用SPSS 20.0软件进行统计分析。正态分布的计量资料以xˉ±S表示,组间比较采用t检验;非正态分布的计量资料以中位数(P25,P75)表示,组间比较采用Wilcoxon符号秩和检验。计数资料采用例数(%)表示,组间比较采用χ2检验或Fisher精确概率法。以基因诊断结果为金标准,血清酶为待检测标准,绘制ROC曲线获取血清酶的界值,绘制四格表计算血清酶诊断DMD的诊断参数。P<0.05为差异有统计学意义。
2 结果
2.1 一般情况 24例PMD患儿纳入分析,均为男孩,2月龄至2岁11月。婴儿组11例,平均年龄(0.6±0.3)岁;幼儿组13例,平均年龄(2.1±0.6)岁。
2.2 基因检测结果 24例患儿及其父母均留取血液样本,并对其父母行DMD基因突变点的验证,母亲是该基因突变携带者19例(79.2%),其中DMD组14例,BMD组5例;新发突变5例(20.8%);两组变异来源差异无统计学意义(P=0.63)。高通量检测DMD基因变异结果显示,大片段缺失15例(62.5%),经典剪切位点变异4例(16.7%),点突变3例(12.5%),大片段重复2例(8.3%)。24例中发生变异的外显子有2个高频突变区域,即位于中央区的第45~55号外显子(17例,70.8%)和5’端第5~18号外显子(4例,16.7%),变异类型包括缺失、重复、剪切位点突变,而所有点突变均在此区域外。DMD组和BMD组突变涉及的外显子分布大致相同(图1)。DMD组18例,平均年龄(1.3±0.9)岁;BMD组6例,平均年龄(1.8±0.8)岁,两组年龄差异无统计学意义(P=0.21)。

图1 PMD患儿基因变异涉及的外显子位置
2.3 临床特征 24例的父母均否认近亲结婚,其中4例(DMD组3例,BMD组1例)有PMD家族史,2例因表哥和亲哥确诊DMD而就诊。22例因其他疾病就诊过程中发现AST、ALT升高,其中13例被诊断为“肝功能不全”行护肝治疗3 d至2年。
23例无肌无力表现,1例(2岁11月)因入院体检发现肝功能异常,护肝治疗效果欠佳,同时发现下肢近端肌力Ⅳ+、Gower征阳性。4例出现腓肠肌肥大、坚硬。婴儿组11例独坐时间均在7~8月龄;幼儿组13例中5例(DMD组4例,BMD 1例)独走时间>18月龄,8例在12~18月龄。
2.4 血清酶检测 表1显示,24例患儿均行AST、ALT、CK、LDH和CK-MB检测,结果均明显升高,且DMD组均高于BMD组,其中ALT、CK、LDH和CK-MB水平两组差异有统计学意义;婴儿组和幼儿组上述指标差异均无统计学意义。依据AST、ALT、CK、LDH和CK-MB在PMD患儿中诊断DMD的价值行ROC曲线分析,分别计算各血清酶学指标的约登指数,以约登指数最大数值时的取值为最佳界值(表2),以ALT的诊断AUC最大,LDH的特异度最高。

表1 PMD患儿不同年龄和诊断的血清酶学比较[M(P25,P75)]
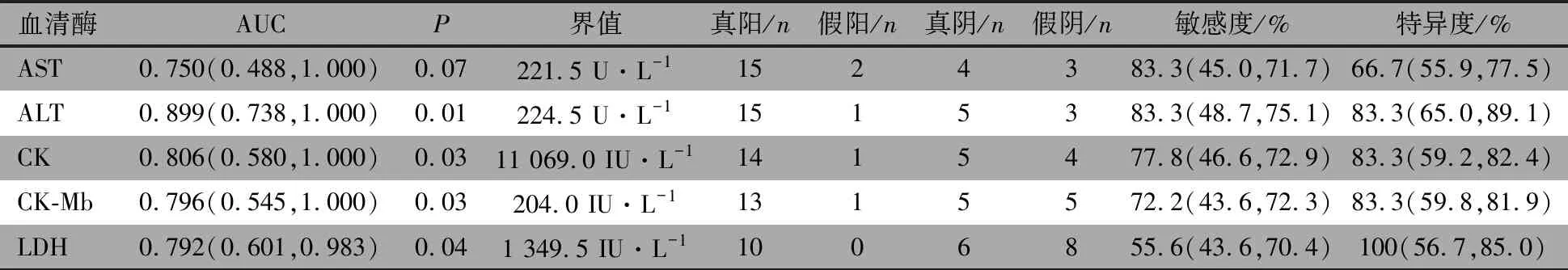
表2 血清酶学指标诊断DMD的ROC曲线结果和最佳界值结果汇总(95%CI)
3 讨论
PMD是由DMD基因突变引起dystrophin蛋白缺陷导致的X连锁隐性遗传的神经肌肉疾病。dystrophin蛋白连接细胞骨架肌动蛋白与细胞外基,主要在骨骼肌中表达。婴幼儿时期的DMD和BMD基本处于症状前期,此时骨骼肌尚处于肌肉代偿期,两者临床症状无表现或较轻微,无明显差别,随着基因治疗技术逐步实现,通过分析症状前期PMD患儿的各项指标,早期区分DMD和BMD并干预,具有重要的临床意义。
本文24例婴幼儿PMD的临床表现无明显差别,早期症状隐匿,无明显肌无力表现,仅1例出现Gower征,可出现如独走时间较晚等运动里程碑延迟,但均有血清酶升高。婴幼儿PMD常因其他疾病行肝功能检测时发现ALT、AST升高,故易误诊为肝脏疾病而延长确诊时间。韩春锡等[6]研究显示患儿就诊原因为CK、AST、ALT升高占79%;罗智强等[7]研究中纳入39例DMD患儿,53.85%因发现肝酶异常而就诊;贺影忠等[2]研究纳入96例DMD患儿,23例(24.0%)误诊为肝炎或肝损害、心肌炎、脑瘫等;李媛媛等[8]报道的24例DMD患儿均有保肝治疗史。本文13例(48.2%)有“肝功能不全”治疗史,最长治疗2年,可能与非专科医生的经验不足有关,亦可能是经护肝治疗致使血清转氨酶降低而造成“治疗有效”的假象。形成该假象的原因为常用的护肝药如还原性谷胱甘肽,具有维持膜稳定性和抑制炎症反应的作用[9,10],而PMD的次级病理机制是dystrophin蛋白缺陷导致细胞膜受损及由此继发的炎症反应[11],病因未去除,转氨酶始终无法恢复至正常水平。婴幼儿血清转氨酶升高尤其同时伴有心肌酶谱增高是早期筛查PMD的重要线索,当患儿出现病因不明且疗效欠佳的“肝炎”、“心肌炎”时应检测转氨酶、CK、LDH等肌肉相关酶学指标,若均升高则应排查是否为PMD等肌病。
多篇文献[4,6-8]报道血清AST、ALT、CK升高是诊断PMD的相关指标,而血清AST、ALT、CK数值与PMD病情并无相关性。有文献报道[12],DMD患儿的血清AST、ALT、CK数值略高于BMD患儿,但两组差异无统计学意义。本文婴幼儿时期的BMD和DMD患儿的血清酶学水平差异有统计学意义,BMD组血清ALT、CK、CK-MB、LDH的数值均明显低于DMD组,可能与纳入的病例均为婴幼儿有关。既往因经济与医疗水平所限,患儿常在症状出现后就诊,DMD于5岁左右出现临床症状,而BMD于11岁左右[4,5],远超过婴幼儿的年龄范围。本文中婴幼儿时期血清酶指标BMD显著低于DMD组,提示血清酶学水平越低,BMD的可能性越大,当ALT>224.5 U·L-1、CK>11 069 IU·L-1、CK-Mb>204 IU·L-1、LDH>1 349.5 IU·L-1时为DMD的可能性更高,尤其是LDH>1 349.5 IU·L-1具有高度特异性,
可为临床早期区分BMD和DMD提供一定依据,尤其对于缺乏基因检测手段及病理活检的基层医疗机构具有重要临床意义。
DMD基因是目前发现的人类最大的基因,约2.3 Mbp,由79个外显子组成,数目庞大且结构复杂,突变率高且突变形式多样[13]。本文24例PMD患儿中,大片段缺失、大片段重复、点突变、经典剪切位点变异分别占62.5%、8.3%、12.5%、16.7%,与文献报道相近[13]。所有类型的突变可发生在基因的任何位置,本文位于中央区的第45~55号外显子、位于5’端第5~18号外显子2个基因突变热点区域,与既往文献报道的突变热点区域基本重合[4]。本文病例所有缺失、重复、剪切位点突变都发生在热点区域,所有的点突变均在热点区域之外,是巧合还是另有原因,还需进一步探究。Zhao等[3]报道121例DMD,对64例进行验证后证实43.8%突变来自母亲。本文病例变异来源于母亲高达79.2%,其中DMD组77.8%(14/18),BMD组85.7%(5/6),两组差异无统计学意义。
综上所述,血清肌酶水平升高为PMD患儿症状前期的主要表现,ALT、CK、CK-MB、LDH对PMD患儿的分组具有指导意义,尤其LDH>1 349.5 IU·L-1对诊断DMD具有高度特异性。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
华人时刊(2019年23期)2019-05-21
中国生殖健康(2018年4期)2018-11-06
健康管理(2017年4期)2017-05-20
妈妈宝宝(2017年4期)2017-02-25
百科知识(2015年18期)2015-09-10
湖北农业科学(2014年11期)2014-09-10
