
靶向EGFR治疗非小细胞肺癌的小分子研究进展
2021-12-13 02:56:52江筱韵李梦玲杨洋李嘉颖杨静雅许芳
药学研究 2021年11期
江筱韵,李梦玲,杨洋,李嘉颖,杨静雅,许芳
(暨南大学药学院,广东 广州 510632)
1 概述
非小细胞肺癌(non-small cell lung cancer,NSCLC)是全球死亡率最高的恶性肿瘤之一[1],在我国城市人口恶性肿瘤死亡高居第一位。70%的NSCLC患者在确诊时已是晚期,5年平均生存率仅约为15%,恶性程度高,严重危害患者生存[2]。其中,肺腺癌(lung adenocarcinoma,LUAD)和肺鳞状细胞癌(lung squamous cell carcinoma,LUSC)是主要的两种细胞分型。吸烟是诱发NSCLC的主要病因。肺癌生物学发病机制的研究进展对NSCLC的有效治疗至关重要。根据患者致病基因的筛查配以相应的治疗方法已成为目前个体化治疗方案的核心。已发现非小细胞肺癌发生的突变包括表皮生长因子受体(epidermal growth factor receptor,EGFR),Kirsten rat sarcoma (KRAS)以及anaplastic lymphoma kinase(ALK)等。其中表皮生长因子受体(epidermal growth factor receptor,EGFR)突变,EGFR调控细胞的增殖、分化及凋亡。通过抑制其酪氨酸激酶的活性表达,可以有效抑制肿瘤的生长和增殖[3-4]。笔者主要总结了非小细胞肺癌发生发展的病理学机制以及靶向EGFR治疗NSCLC的小分子研究进展。
1.1 EGFR及其相关耐药性突变 EGFR是一种受体型酪氨酸激酶,由胞外区(N末端)、单次跨膜区和胞内区(C末端)组成。EGFR胞内区的酪氨酸激酶域有ATP结合位点,能催化C末端氨基酸残基以实现自身磷酸化。作用于EGFR的配体主要有:表皮生长因子(epidermal growth factor,EGF)、转化生长因子α(transforming growth factor α,TGFα)和调节素(amphiregulin,AR)。EGFR与配体结合后发生同源或异源二聚,进一步引起自身磷酸化,磷酸化的EGFR可调控下游蛋白从而启动不同的信号转导通路。EGFR主要调控的信号通路有:Ras/Raf/MAPK、PI3K/Akt和JAK/STAT。将信号由胞质传递到胞核,分别调控细胞增殖,分化及凋亡等一系列生理过程。EGFR基因家族表达于正常人体上皮组织中,如皮肤、毛囊、胃肠道壁等,对细胞生长,分化,组织更新和伤口愈合起重要作用[5-6]。
国内外先后开发了多个EGFR抑制剂用于临床。一代EGFR抑制剂,代表药物包括:阿斯利康公司(AstraZeneca)推出的吉非替尼(易瑞沙,Iressa)和罗氏制药(Roche)的厄洛替尼(特罗凯,Tarceva)[7-8]。NSCLC患者用药后缓解率、生活质量和无进展生存期均明显改善。然而,患者在连续药物10-12个月后,大部分产生了耐药性[9]。二代EGFR抑制剂如勃林格殷格翰公司(Boehringer Ingelheim)的阿法替尼(Afatinib)、辉瑞公司(Pfizer)的达克替尼(Dacomtinib)和惠氏制药(Wyeth)开发的来那替尼(Neratinib)等[10-11]。上述药物由于选择性差,药物毒副作用(严重皮疹和腹泻)呈剂量依赖性,以及体内和临床试验中产生EGFR的 T790M位点突变诱发耐药等问题而逐渐退出临床[12-13]。第三代抑制剂是EGFR T790M选择性非可逆抑制剂,以阿斯利康(AstraZeneca)公司开发的奥希替尼(泰瑞莎,Osimertinib,AZD9291或TAGRISSOTM),克洛维斯肿瘤公司(Clovis Oncology)的Rociletinib(CO-1686,AVL-301),勃林格殷格翰公司的Olmutinib(HM61713),诺华制药公司(Novartis)的Nazartinib(EGF816)和安斯泰公司(Astellas)的ASP8273等为代表性药物[13-14],具体见图1。上述药物适用于治疗晚期NSCLC患者,特别是携带EGFR常见突变Del19和L858R,以及T790M突变的患者。其中,Osimertinib和Rociletinib对EGFR T790M相对于EGFR WT型的选择性分别为40倍和14倍。上述两种药物分别于2013年和2014年获得美国FDA孤儿药认证,并相继获得了美国FDA“突破性治疗药物资格”。然而用药一段时间后,NSCLC患者相继出现新复发耐药,其主要原因与EGFR发生了新的耐药突变有关[15-16]。当前国内外围绕克服Osimertinib(Osi)为代表的三代抑制剂耐药做了大量研究工作,本文主要总结和归纳了近年来NSCLC领域Osimertinib介导的EGFR主要耐药性突变,以及为克服Osimertinib而开发的小分子(抑制剂和降解剂)。以期为开发克服EGFR突变耐药的NSCLC药物提供研究基础。
2 EGFR突变介导Osimertinib耐药机制
鉴于NSCLC存在靶点多种突变型特征,其治疗已进入分型治疗阶段,即:根据不同的分子特征来选择相应的分子靶向药物进行治疗,可有效提高患者的中位生存时间。EGFR小分子抑制剂是治疗NSCLC患者的有效策略,然而治疗后患者往往会出现耐药,如何解决耐药,是当前临床亟待克服的世界难题。引起EGFR抑制剂耐药的主要原因是EGFR20号外显子出现T790M点突变,临床上该位点突变的NSCLC患者超过50%。三代抑制剂为克服这一突变而诞生。

图1 已获批上市的三代EGFR抑制剂药物
典型代表性药物是Osimertinib又名Tagrisso或AZD9291,中文名:奥西替尼(泰瑞沙)(见图1)。可用于治疗EGFR T790M突变阳性的局部晚期或转移性非小细胞肺癌,疗效显著,且副作用较低。2013年获得美国FDA孤儿药认证,并获得了美国FDA“突破性治疗药物资格”。由于可通过血脑屏障,对脑转移也显示有一定疗效。2015年11月13日经FDA加速批准上市,是首个获批上市用于经EGFR-TKI治疗失败后病情进展的T790M突变阳性NSCLC靶向药物。2018年4月,美国FDA进一步批准了Osimertinib作为治疗携带EGFR L858R、EGFR del E746-A750等激活性突变的转移性NSCLC的一线药物,大大扩展了其临床使用范围。值得关注的是,奥希替尼与EGFR不可逆结合,不仅可以克服由于一二代EGFR-TKI耐药的EGFR T790M突变,同样可用于治疗EGFR敏感突变(19Del和21L858R)的晚期NSCLC患者[17]。围绕奥希替尼开展的系列临床多中心开放性Ⅰ/Ⅱ期研究工作(简称AURA)将I期和拓展性研究包括起来,得出更重要的结论。
AURA[18-20]系列临床研究中,奥希替尼最常见的不良事件包括皮疹、腹泻、皮肤干燥和甲沟炎等。此外相比其他一二代EGFR-TKI,奥希替尼3级及以上不良事件发生率较低(32% vs 41%)。一项奥希替尼AURA ex和AURA2[19]的日本人群的合并分析显示,ILD(interstitial lung disease,间质性肺疾病)的发病率约为6.2%,提示日本人群的ILD发病率拉高整体人群的ILD发病率。而在FLAURA[21]和AURA的系列研究中,共纳入284名中国患者,其ILD的发生率为1.4%,因为ILD导致死亡的比例为0%。
然而,随着EGFR三代抑制剂Osimertinib的广泛应用,EGFR T790M获得性耐药突将临床治疗NSCLC带入新困境[22]。Osimertinib的耐药机制复杂,主要包括:①EGFR发生Cys797Ser797突变(EGFRC797S),使得药物无法与蛋白形成共价非可逆结合,降低药物与ATP的竞争活性;②非小细胞肺癌向小细胞肺癌转化;③促癌基因,如MET、k-Ras、ErbB-2、EphA2、FGFR、TAM受体成员Mer和Axl、NF-κB、B-Raf和PIK3CA等,发生突变和增强[23]。其耐药机制主要可分为EGFR依赖型和EGFR非依赖型(见图2)。

图2 EGFR突变型NSCLC潜在治疗策略及耐药机制示意图
2.1 EGFR依赖型耐药机制 靶向治疗策略的常见耐药机制之一为靶点的获得性突变。在部分奥希替尼用药的NSCLC患者中。后续发生EGFR C797S突变阻断了奥希替尼与靶点EGFR C797位点的有效结合[24],从而使得Osi失效。此外,也在Osi耐药的患者中发现发现EGFR G796D、G796S/R、L792F/Y/H、C797G、L718Q等继发性EGFR突变现象[25]。此外在Osi耐药患者中也发现EGFR 外显子19和野生型显著增强,以及EGFR T790M缺失的现象。以及EGFR受体蛋白总体表达下降。然而有趣的是,EGFR生长因子在Osi耐药后病情发生恶性进展的NSCLC患者中表达水平持续升高。这些现象反映了Osi耐药机制的复杂性。
2.2 EGFR非依赖型耐药机制 已知许多受体酪氨酸激酶和EGFR具有相似的下游调节途径,从而激活肿瘤恶性进展[17]。NSCLC细胞内,可通过旁路激活的方式调节其他激酶活性,从而激活EGFR下游信号通路,“逃逸”Osi对EGFR下游信号的抑制,促进细胞存活。这一旁路激活方式包括了一系列非EGFR激酶的突变性激活,增强或缺失。如BRAF V600E、PIK3C E545K、PTEN Y27C、CTNNB1 S37F和TSC2 N486I突变;KRAS G12S突变,NRAS E63K和Q61K突变。Src家族激酶激活,YES1增强以及MAPK1、AKT3和AXL异常过表达。PTEN缺失的现象也在部分Osi耐药患者中发现。
此外,一些疾病表型的变化也在部分Osi耐药患者中观察到。如部分Osi耐药患者表现为非小细胞癌向小细胞癌(SCLC)的组织学转化和上皮间质转化(EMT),这也表明了Osi耐药机制的复杂性。
3 第四代EGFR小分子抑制剂
3.1 EAI045 EAI045(见图3)是一种变构抑制剂,它靶向选择产生耐药的EGFR突变体,但不作用于野生型EGFR受体。对EAI045的细胞活性的初步研究表明,它在L1975R/T790M突变型NSCLC细胞系H1975细胞、用L858R/T790M突变体稳定转染的NIH-3T3细胞中能有效降低EGFR自磷酸化,但不能完全消除。进一步研究发现,对一组EGFR突变的Ba/F3细胞,EAI045能抑制L858R/T790M和L858R突变细胞的增殖,但不抑制exon19del/T790M或亲代Ba/F3细胞的增殖,这表明其对靶标的突变体具有选择性。后续研究表明,EAI045与西妥昔单抗联合用药在发生EGFR(L858R/T790M)和EGFR(L858R/T790M/C797S)驱动突变的肺癌小鼠试验中证明有效[26]。
3.2 JBJ-04-125-02 To等[27]研究发现,JBJ-04-125-02(见图3)在EGFR C797S模型中在体内和体外均可作为单一药物有效。JBJ-04-125-02能抑制用EGFR L858R,L858R/T790M或L858R/T790M/C797S突变稳定转染的Ba/F3细胞系中细胞的增殖能力,而不抑制亲代Ba/F3或野生型EGFR Ba/F3细胞的生长,其抑制上述3种突变的IC50分别为1.0 nmol·L-1、0.4~0.5 nmol·L-1、0.0~0.1 nmol·L-1。值得注意的是,JBJ-04-125-02是唯一可以抑制细胞增殖的单一药物化合物,其联合西妥昔单抗使用时,相比于EAI045也是最有效的。继而进行的体内药效学研究表明,在EGFR L858R/T790M/C797S基因工程小鼠体内,JBJ-04-125-02治疗在治疗4周内导致明显的肿瘤消退。
3.3 2,9-二取代8-苯硫基-9H-嘌呤化合物 Hei等[28]合成了31个包含2,9-二取代8-苯硫基/苯亚磺酰基-9H-嘌呤支架的新型EGFR抑制剂,并进行了生物学评估。其中,化合物C9(图3)对HCC827细胞株的IC50为29.4 nmol·L-1,对EGFR L858R的IC50为1.9 nmol·L-1。化合物C12对EGFR L858R/T790M/C797S表现出中等抑制活性(IC50=114 nmol·L-1)。Western blot分析表明,C9可以显著抑制EGFR磷酸化。其后进行体内测试发现,化合物C9在已建立的裸鼠HCC827移植瘤模型中,口服给药5.0 mg·kg-1能明显抑制肿瘤生长。这些结果表明2,9-二取代8-苯硫基/苯基亚磺酰基-9H-嘌呤化合物可以作为有效的EGFR(L858R)抑制剂和抗癌药。此外,优化化合物C12可能可以发现新的第四代EGFR-TKI。
3.4 4-氨基吡唑嘧啶类化合物 Engel等[29]设计了一类4-氨基吡唑并嘧啶的核心结构的化合物,作为新型EGFR T790M突变抑制剂,合成了1a~1c 3个化合物(见图3)。整个系列的化合物对H1975细胞(GI50值分别为0.21、0.49和0.14 μmol·L-1,分别对应1a、1b和1c)有良好的抑制作用,同时对EGFR野生型(WT)细胞(A431;26~49倍)的抑制作用降低。进一步研究发现,1c对获得性耐药性L858R/T790M/C797S有中等抑制作用(IC50为88 nmol·L-1),其亲和力约是1a、1b的20倍。这一结果提示4-氨基吡唑嘧啶类化合物可能刺激针对获得性T790M和C797S耐药的第四代EGFR抑制剂的发展。

图3 第四代EGFR抑制剂(上)
3.5 三取代咪唑类化合物 Günther等[30]基于高效可逆p38抑制剂的设计出了三取代咪唑类化合物,该类化合物能够有效抑制EGFR L858R/T790M/C797S三重突变体。生物实验表明,化合物9(见图4)在双突变EGFR L858R/T790M酶测定中,IC50值为18 nmol·L-1。化合物19a(见图4)是一种高效的EGFR抑制剂,针对EGFR-L858R/T790M测定的细胞比野生型具有约300倍高的选择性,且对所有三种激酶变异体显示IC50值低于1 nmol·L-1。在此基础上设计合成的共价化合物31a以及可逆对应物31b(见图4),在1 nmol·L-1以下的生化环境中表现出对EGFR-L858R/T790M/C797S三重突变体强烈的抑制作用。这些研究表明了三取代咪唑类化合物有希望作为三代后的新一代EGFR抑制剂克服三重突变的可能性。
3.6 吡咯嘧啶类化合物 Lategahn等[31]设计了基于吡咯嘧啶支架的耐药守门突变体EGFR-L858R/T790M的共价抑制剂。该研究显示N型烷基化吡咯嘧啶类化合物29a-l和O型烷基化吡咯嘧啶类化合物19a~h有较高的抗EGFR活性,其中化合物19g和19h(见图4)的活性最高,IC50值均约为9 nmol·L-1。与前三代EGFR-TKIs相比,这两类化合物具有更好的生化效力。因此,吡咯嘧啶类化合物可作为新一代EGFR抑制剂的研究方向。
4 新兴EGFR小分子降解剂
分子靶向药物出现耐药几乎是无法避免的,有些新的突变位点可能会难以靶向而造成新一代药物研究开发困难。近年来,新兴的蛋白水解靶向嵌合体(proteolysis-targeting chimeras,PROTACs)技术能精准降解靶蛋白,使以往“难以靶向”的靶点的药物研发成为可能,是一种全新的抗癌策略。PROTAC(靶向蛋白降解技术)是一种双功能小分子,其一端是结合靶蛋白(protein of interest,POI)的配体,另一端是结合E3泛素连接酶的配体,二者之间通过一段linker连接。这个三元复合物形成后,在体内可以将靶蛋白和E3酶拉近,使靶蛋白被打上泛素标签,最后通过泛素-蛋白酶体途径(ubiquitin proteasome system,UPS)降解[32]。在蛋白质被降解后,PROTAC分子可以被释放并且进入下一个降解过程(见图5)[33]。
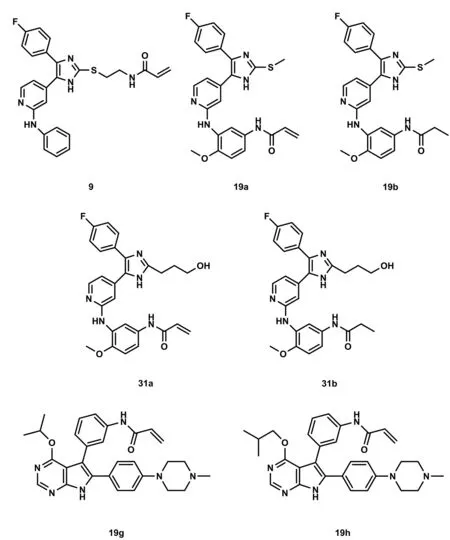
图4 第四代EGFR抑制剂(下)

图5 proteolysis targeting chimera(PROTAC)技术
迄今为止,尽管在人体内发现的E3连接酶有600多种,但已报道的可以用于PROTAC的相关E3泛素连接酶只有少数的几种,如:CRBN(cereblon)、VHL(vonhippel-lindau)、MDM2(mouse double minute 2)、IAPs(inhibitor of apoptosis proteins)等[34]。
4.1 EGFR PROTAC研究进展 在治疗非小细胞肺癌(NSCLC)的过程中,第一代和第二代的EGFR抑制剂出现了耐药性,第三代奥希替尼等EGFR抑制剂虽然克服了第一代和第二代EGFR抑制剂的耐药问题,但是干扰EGFR不可逆抑制剂与EGFR之间的共价结合[35-36]。因此,开发EGFR降解剂正面临着挑战。
幸运的是,2018年Crews团队首次证明了PROTACs能够降解受体酪氨酸激酶(receptor tyrosine kinase,RTKs),并且报道了一种以EGFR抑制剂Afatinib为EGFRL858R/T790M蛋白亲和配体,以VHL为E3连接酶配体的双分子靶向蛋白降解剂(PROTAC3,见图6)[37]。同时,他们还提供了基于相应抑制剂的3种RTKs(EGFR、 HER2、 c-Met)的PROTAC成功降解实例,其中包括EGFR和c-Met的多个突变体。这个研究极大地扩展了跨膜蛋白可以作为PROTACs的潜在靶点,并提出了EGFR-PROTACs可以作为克服C797S介导的耐药性的潜在策略的新想法。最近,国内Jin Jian课题组以Gefinitib为头部分子设计并得到了MS39和MS154两个EGFR降解剂小分子,通过实验证明这两个小分子能有效降低EGFR突变体的蛋白水平并抑制肺癌细胞中的下游信号传导[38];张三奇团队设计并证明了一个2,4,6-三取代嘧啶[3,4-d]嘧啶衍生物可以作为EGFRDel19的降解物。其中,PROTACs2和10能够诱导HCC827中的EGFR降解,(DC50值分别为34.8和45.2 nmol·L-3),且降解剂2和10可显著诱导HCC827细胞凋亡,并在G1期阻滞细胞(见图6)。这些发现表明,在HCC827细胞中EGFEDel19可以被所设计的降解剂有效地靶向降解[39]。同年,丁克团队在XTF-262的基础上设计合成了一个化合物14O可有效和选择性地降解EGFRL858R/T790M(DC50值为5.9 nmol·L-3),且对野生型蛋白质无影响,他们使用的E3泛素连接酶部分是VHL配体(见图6)。进一步的研究机理表明,降解过程是通过泛素-蛋白酶体途径介导的,因此,化合物14O可作为新的EGFRL858R/T790M降解剂的起始先导分子[40]。
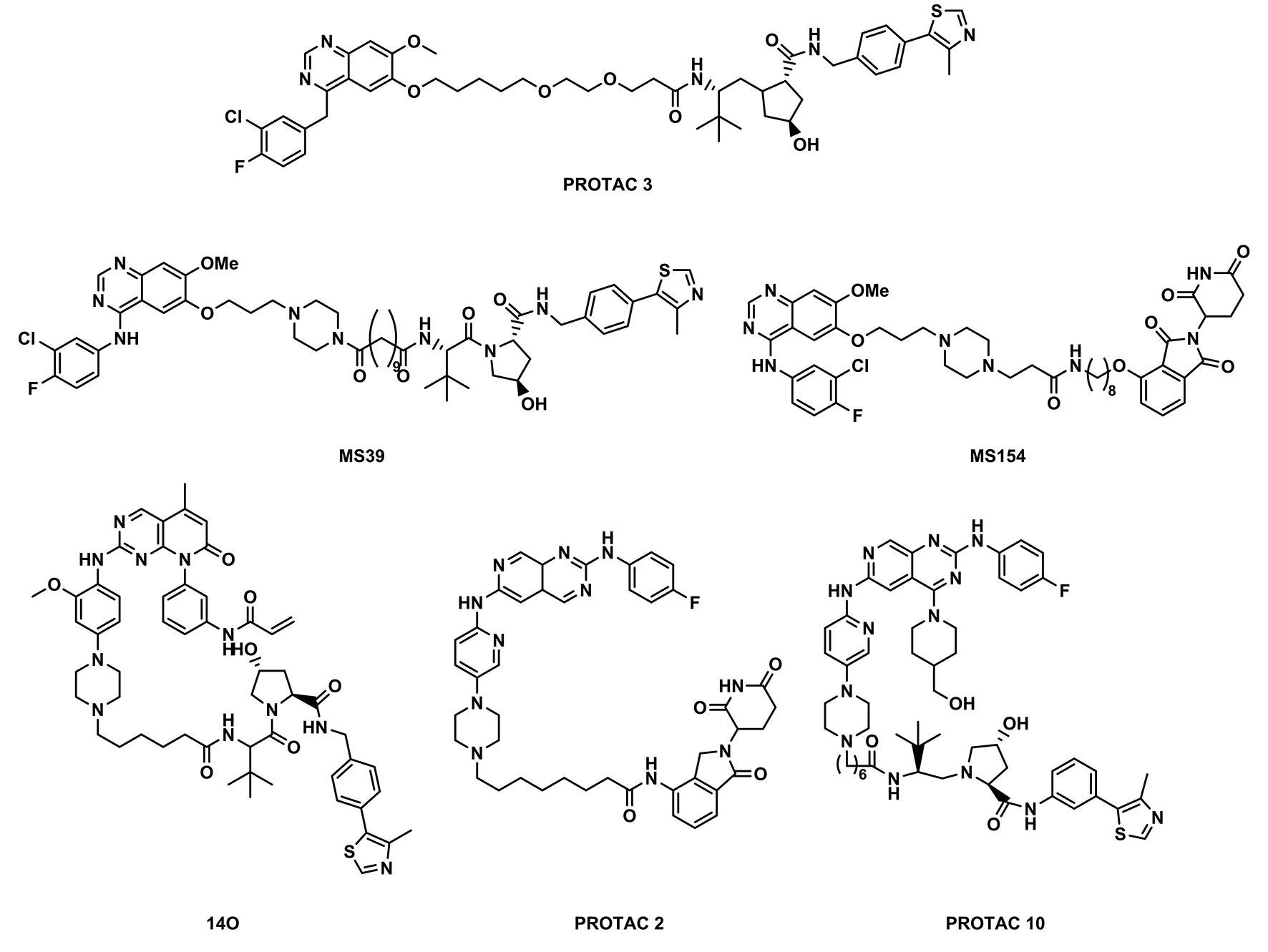
图6 EGFR小分子降解剂
5 总结与展望
靶向小分子发生耐药是临床NSCLC治疗难题,奥希替尼能有效克服EGFR T790M突变耐药在临床引起广泛关注。然而,随着EGFR三代抑制剂Osimertinib的广泛应用,EGFR T790M获得性耐药突变将临床治疗NSCLC带入新困境[22]。Osimertinib的耐药机制复杂,既有EGFR依赖的,也有EGFR非依赖的机制。主要包括:①EGFR发生Cys797 Ser797突变(EGFR C797S),使得药物无法与蛋白形成共价非可逆结合,降低药物与ATP的竞争活性;②非小细胞肺癌向小细胞肺癌转化;③促癌基因,如MET、k-Ras、ErbB-2、EphA2、FGFR、TAM受体成员Mer和Axl、NF-κB、B-Raf和PIK3CA等,发生突变和增强。尽管应对Osimertinib耐药的第四代EGFR抑制剂相继开发,也取得了一定疗效。但针对靶点开展的小分子占位驱动型研发不可避免地会带来靶点突变而耐药。新兴PROTAC技术为事件驱动型疗法,以清除致病靶蛋白为目标,而对其他蛋白不降解。EGFR T790M小分子降解剂只降解肿瘤组织中的EGFR T790M突变型蛋白,而对正常组织中的EGFR不降解。这在很大程度上避免了副作用。近年来,PROTAC技术发展迅速,利用PROTAC技术治疗EGFR突变的非小细胞肺癌的研究也正在进行当中,虽然在其发展过程中面临E3酶配体较少等问题,但这项技术仍为克服Osimertinib耐药提供了重要解决方案。
猜你喜欢
保健医苑(2022年5期)2022-06-10 07:46:38
中学生数理化·中考版(2021年12期)2021-12-31 03:24:40
中学生数理化·中考版(2021年11期)2021-12-06 07:29:16
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
中学化学(2017年6期)2017-10-16 17:22:41
上海农业学报(2017年3期)2017-04-10 12:39:26
医学研究杂志(2015年7期)2015-06-22 11:01:01
中国当代医药(2015年16期)2015-03-01 02:03:13
中国药理学通报(2014年2期)2014-05-09 08:22:39
