
信阳10号叶绿体基因组及其系统进化
2021-12-11 01:59:22闫明慧刘柯王满吕颖张倩
茶叶科学 2021年6期
闫明慧,刘柯,王满,吕颖,张倩
信阳10号叶绿体基因组及其系统进化
闫明慧1,刘柯2,王满2,吕颖1,张倩1
1. 信阳师范学院生命科学学院/河南省茶树生物学重点实验室,河南 信阳 464000;2. 信阳师范学院国教学院,河南 信阳 464000
信阳10号是适制信阳毛尖的国家级良种,然而其起源以及与其他茶树品种之间的进化关系尚不清晰。利用MGI2000平台对信阳10号进行测序,组装获得了信阳10号的完整叶绿体基因组并对其结构进行分析,同时,为探究信阳10号与其他茶树的进化关系,构建了46个物种的叶绿体基因组系统发育树。结果表明:(1)信阳10号叶绿体基因组大小为157 041 bp,包括2个反向重复区(IR,26 078 bp),1个大单拷贝区(LSC,86 594 bp)和1个小单拷贝区(SSC,18 291 bp);共注释得到叶绿体基因113个,包括79个蛋白质编码基因,30个tRNA基因和4个rRNA基因。(2)在信阳10号的叶绿体基因组中共检测到了74个SSR位点,大部分SSR由A/T组成。(3)贝叶斯法构建的系统进化关系树显示,信阳10号与福建铁罗汉关系最近,并且两者的叶绿体基因组完全相同,推测可能来源于相同母本;信阳10号与韩国茶Chamnok和Sangmok、福建白鸡冠、云南德宏茶也有较近的亲缘关系。研究结果为进一步探究茶树起源与演化以及分子育种提供了基础。
茶树;信阳10号;叶绿体基因组;系统发育分析
茶树[(L.) O. Kuntze]是一种自交不亲和的多年生木本植物,种间频繁杂交导致基因组杂合度高(2.8%),同时核基因组较大(约3.0 Gb)且重复序列含量极高[1],很难筛选出有效的核基因组数据来区分种间关系。以往的研究多利用随机扩增序列的多态性(Randomly amplified polymorphic DNA,RAPD)作为分子标记探究茶树品种或茶属内部的系统进化关系[2-5]。
相对于核基因组,叶绿体基因组多为母系遗传,其进化路线独立、进化速率适中、核苷酸替换率较低、基因结构稳定,在系统研究方面易于提供物种进化信息[6]。叶绿体基因组的基因密度高于线粒体基因组和核基因组,除去基因间区、内含子和调控区,茶树叶绿体基因组的基因区约占整个叶绿体基因组核苷酸序列的50%[7]。此外,叶绿体基因组具有拷贝数量较大的特点,有助于基因序列SNP的识别,为种间变异分析、系统重建和种群遗传分析提供信息[8]。
叶绿体基因组一般为典型的四分体结构,即包含一段大单拷贝区(Large single copy region,LSC),一段小单拷贝区(Small single copy region,SSC),以及将这两段分开的序列相同、方向相反的两个反向重复区(Inverted repeat region,IRA和IRB),基因组大小为170~280 kb[9]。随着测序技术的不断发展,叶绿体基因组数据库也日益充实,目前在NCBI上面已有23个茶树叶绿体全基因组(表1),其中20个分布于中国[10-13]。
利用叶绿体基因组探究茶树系统进化关系的研究陆续开展[14-16]。例如,对6种山茶属的叶绿体基因组进行比对,发现了长期进化过程中不同环境对不同茶树叶绿体基因组的影响,表明叶绿体基因组可以用于区分山茶属的种间关系[15]。Li等[11]为探究武夷茶的起源及进化,对大红袍(cv. Dahongpao)的叶绿体基因组进行测序,构建叶绿体系统进化树,发现龙井(cv. Longjing)与大红袍关系最近,为武夷茶起源提供了依据。
信阳毛尖又称豫毛峰,是中国十大名茶之一,主要产自信阳市浉河区车云山、集云山、云雾山、天云山、连云山、黑龙潭、白龙潭、何家寨[17]。信阳10号(cv. Xinyang 10)是由河南省信阳茶叶试验站于1976—1988年从信阳群体中育成的茶树品种,其主要产地在信阳市浉河区。信阳10号植株中等,分枝匀称、密度大,叶长椭圆形,富光泽,芽叶淡绿色,生育力强,发芽整齐,产量高,抗寒性高于福鼎大白茶[18],多用于制作信阳毛尖。但对于信阳10号的进化和发展历程,以及与其他茶树品种之间的进化关系尚不清晰。
本研究报道了信阳10号茶树叶绿体基因组的完整结构,并与其他茶树品种的叶绿体基因组结构进行比较,通过构建进化树揭示信阳10号与其他茶树品种的进化关系及其在山茶属系统发育中的地位,为茶树品种资源的鉴定、开发以及茶树育种提供理论依据。
1 材料和方法
1.1 试验材料
信阳10号植株采自河南省信阳市浉河区信阳师范学院茶树试验田(N32°8′18.42″,E114°2′5.51″),采摘其新鲜幼嫩叶片,放入–80℃超低温冰箱保存备用。
1.2 试验方法
1.2.1 基因组DNA提取及测序
基因组DNA提取及测序采用LEAGENE植物DNA提取试剂盒(北京雷根生物技术有限公司,EN0206)提取信阳10号基因组总DNA,通过琼脂糖凝胶电泳检测DNA的纯度和浓度,符合测序要求后,送测序公司用MGI2000平台进行PE150双端测序。
1.2.2 叶绿体基因组组装和注释
测序完成后得到的原始序列(Raw reads)先用NGS QC Tool-Kit过滤去除接头及两端低质量序列,得到高质量待分析序列(Clean reads)。再用Bowtie 2[19]软件从过滤数据中筛选出叶绿体基因组的reads,然后使用Unicycler[20]软件对筛选出来的reads进行组装得到contig。最后,以龙井43的叶绿体基因组(GenBank登录号:KF562708)为参考序列,在Geneious 9.0.2软件中拼接contigs,得到完整的信阳10号叶绿体基因组。使用DOGMA在线工具对叶绿体基因组蛋白质编码基因、转移RNA(tRNA)基因和核糖体RNA(rRNA)基因进行预测[21],并利用PGA软件对基因进行功能注释,利用OGDRAW 1.3.1在线软件[22]绘制信阳10号叶绿体基因组图谱。
1.2.3 叶绿体基因组特征分析
利用MISA在线软件[23]鉴定信阳10号叶绿体基因组中的简单重复序列(Simple sequence repeat,SSR),搜索参数设置为:含有完全重复的单核苷酸最小重复数为10,二核苷酸最小重复数为5,三核苷酸最小重复数为6,四、五、六核苷酸最小重复数为3;另外设置2个SSR之间的最小距离为100 bp,如果距离小于100 bp,则2个SSR被当做1个复合微卫星。使用DNASP 6进行滑动窗口分析来计算包括信阳10号在内的17种不同茶树代表品种的叶绿体基因组之间的核苷酸多样性指数(),窗口长度设为800 bp,步长设为200 bp[24]。
1.2.4 系统发育分析
从NCBI数据库下载已公布的16个具有代表性的茶树品种(未选择6个野生大叶茶和1个韩国茶品种Kuntze)和28个山茶属其他物种共44个完整的叶绿体基因组序列,下载近缘的山茶科圆籽荷()作为外类群。用MAFFT软件比对所有叶绿体基因组序列,结果经手工检查与调整后采用贝叶斯法(Bayesian analysis)对系统进化关系进行分析。贝叶斯系统发育树使用MrBayes 3.2.7[25]软件生成,设置剪辑替换模型为GTR,运行2 000 000次,每隔1 000运算取样一次。最后,用Figtree 1.4.2软件对建树结果进行显示和编辑。
2 结果与分析
2.1 基因组基本特征
利用高通量测序,共获得63.4 Gb信阳10号全基因组数据,过滤后得到2.92 Gb叶绿体基因组reads,占全基因组测序数据的4.61%。经过组装,信阳10号叶绿体基因组与大多数被子植物叶绿体基因组一样,为共价闭合的双链环状分子(图1),全长157 041 bp,包括1对IR区(26 078 bp),1个LSC区(86 594 bp)和1个SSC区(18 291 bp),编码区占58%,非编码区占42%。全基因组的GC含量为37.29%,IR区GC含量最高为42.94%,LSC(35.31%)和SSC(30.53%)区相对较低(表2)。信阳10号叶绿体基因组共编码113个基因,其中蛋白质编码基因79个,tRNA基因30个,rRNA基因4个。LSC区包含基因数最多,含有60个蛋白编码基因和21个tRNA基因;SSC区包含了1个tRNA基因()和11个蛋白编码基因;IR区共有19个基因,包含了所有的rRNA基因、8个tRNA基因和8个蛋白编码基因(图1)。与光合作用有关的基因有47个,分别为7个光合系统Ⅰ基因、15个光合系统Ⅱ基因、6个ATP合成酶基因、6个细胞色素复合物编码基因、11个NADH脱氢酶基因、1个二磷酸核酮糖羧化酶大亚基基因和1个依赖ATP蛋白酶单元p基因;与转录和翻译有关的基因包含了4个RNA聚合酶亚基基因、9个核糖体大亚基基因和12个核糖体小亚基基因;此外还有7个其他功能基因(表3)。完整的信阳10号叶绿体基因组及其注释信息已提交至NCBI,登录号为MZ153237。
2.2 SSR分析
SSR广泛分布于叶绿体基因组中。在信阳10号叶绿体基因组中共检测到74个SSR,其中单核苷SSR最多,共56个,均为A/T重复;二核苷酸SSR 4个,为AT/AT重复;三核苷酸SSR 1个,为AAG/CTT重复;四核苷酸SSR 11个,具有5种重复类型,分别是ACAG/CTGT重复(1个)、AAAG/CTTT重复(2个)、AGAT/ATCT重复(3个)、AAAT/ATTT重复(3个)和AGGG/CCCT重复(2个);六核苷酸重复SSR有2个,均为AAAAAG/CTTTTT重复;未检测到五核苷酸SSR。所检测的SSR以A/T重复为主,占所有SSR的75.68%,表明信阳10号叶绿体SSR偏好使用A和T碱基(表4)。
2.3 信阳10号与代表性茶树叶绿体基因组的比较分析
将信阳10号的叶绿体基因组与其他已报道的茶树叶绿体基因组进行比较,发现不同茶树品种间叶绿体基因组大小差异较大,其中印度大叶茶的叶绿体基因组最大,为157 353 bp,韩国Sangmok的最小,为153 044 bp,两者相差接近4 kb。信阳10号的叶绿体基因组大小与福建铁罗汉相同,且碱基差异为0(表5),说明这两个茶树品种具有完全相同的叶绿体基因组序列。
对信阳10号、龙井43、安化茶、云抗10号和大红袍5个茶树品种叶绿体基因组的反向重复区和单拷贝区的边界进行比较,发现不同茶树品种叶绿体基因组区域边界具有高度的保守性(图2)。5个茶树品种的LSC/IRB边界均位于基因内部,距离该基因最后一个碱基46 bp,IRA/LSC边界均位于基因前两个碱基处。5个品种的SSC/IRA边界均位于基因内部,龙井43、云抗10号、大红袍的SSC/IRA边界均位于基因最后一个碱基上游1 069 bp处;信阳10号与之相比有9个碱基差异,位于1 060 bp处,安化茶位于1 049 bp处。IRB/SSC边界位于基因上游,此边界位置变异较多,仅信阳10号与云抗10号相同,位于基因上游57 bp处。

表2 信阳10号叶绿体基因组基本特征
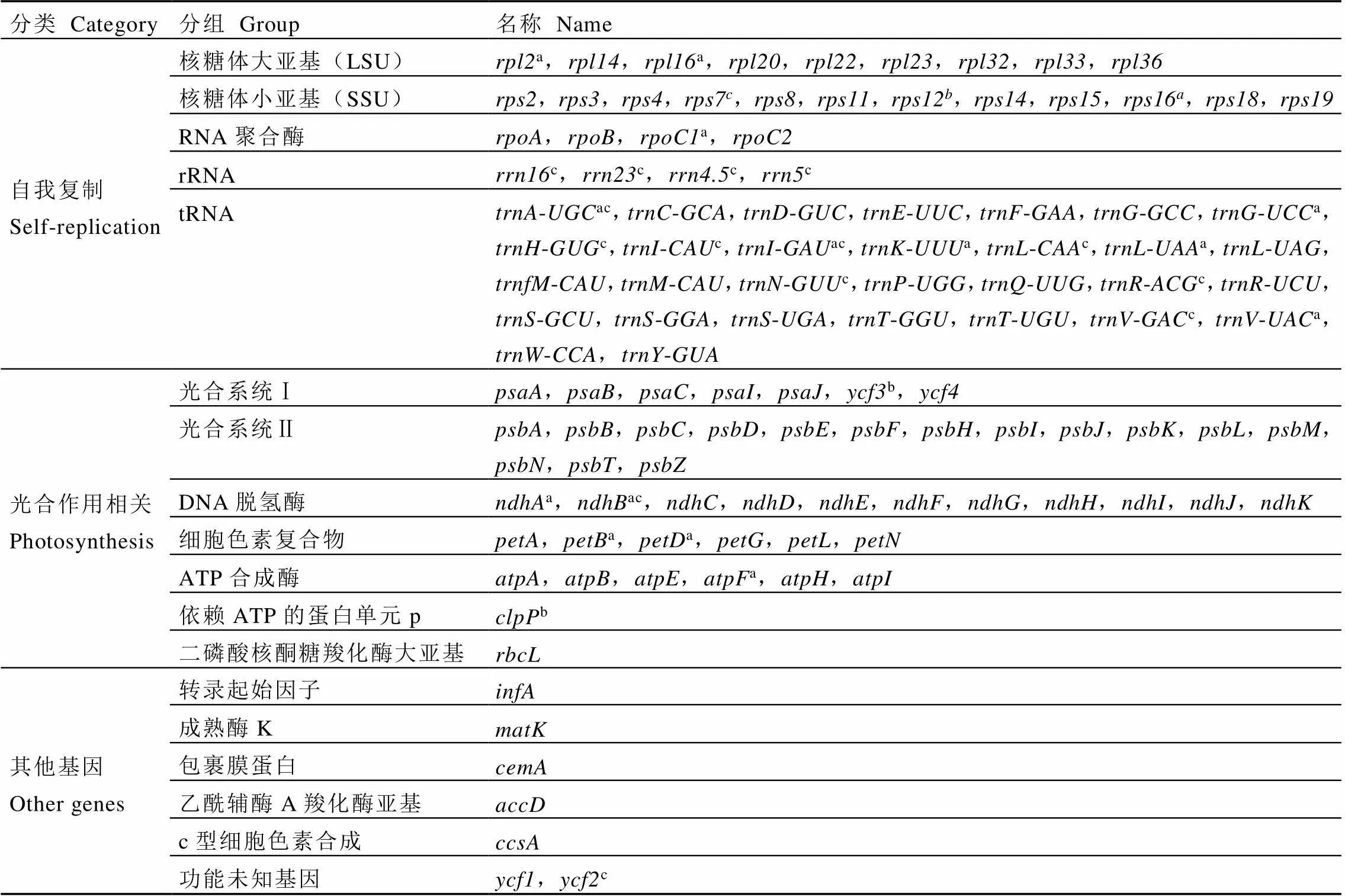
表3 信阳10号叶绿体基因组基因列表
注:上标a和b分别表示基因含有1个和2个内含子;c表示含有2个拷贝基因
Note: Superscript a and b present 1 and 2 introns in protein-coding genes respectively. Superscript c presents 2 copies of genes
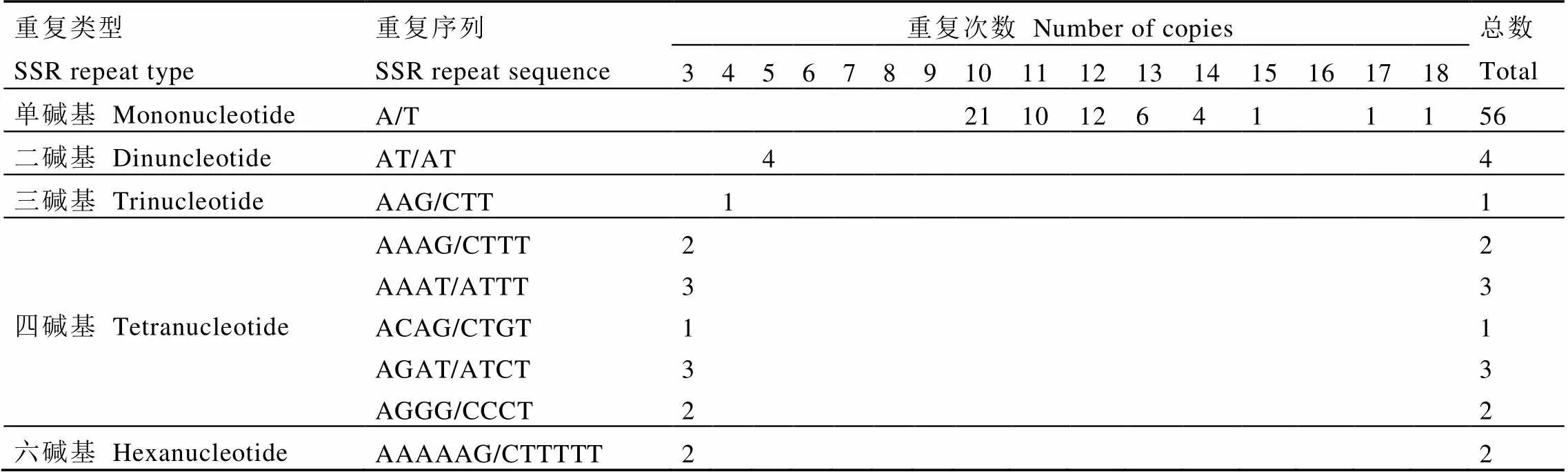
表4 信阳10号叶绿体基因组SSR信息
通过滑动窗口分析,发现17个茶树品种的核苷酸多样性分出了高低差异较大的两个区域(图3)。叶绿体基因组的核苷酸多样性大部分处于较低水平,此区域的核苷酸多样性平均值为0.001 35,而在SSC区(112 750~131 031 bp)突然提高至0.058 08。茶树品种之间叶绿体基因组大部分核苷酸多态性较低的原因可能是茶树同种之间分化程度不高。对于SSC区核苷酸多态性升高的原因,通过多序列比对分析发现,铁观音的叶绿体基因组在SSC区变异程度较大,提高了此区域整体的核苷酸多样性,这个区域共包含了从到功能基因共14个基因(图1)。
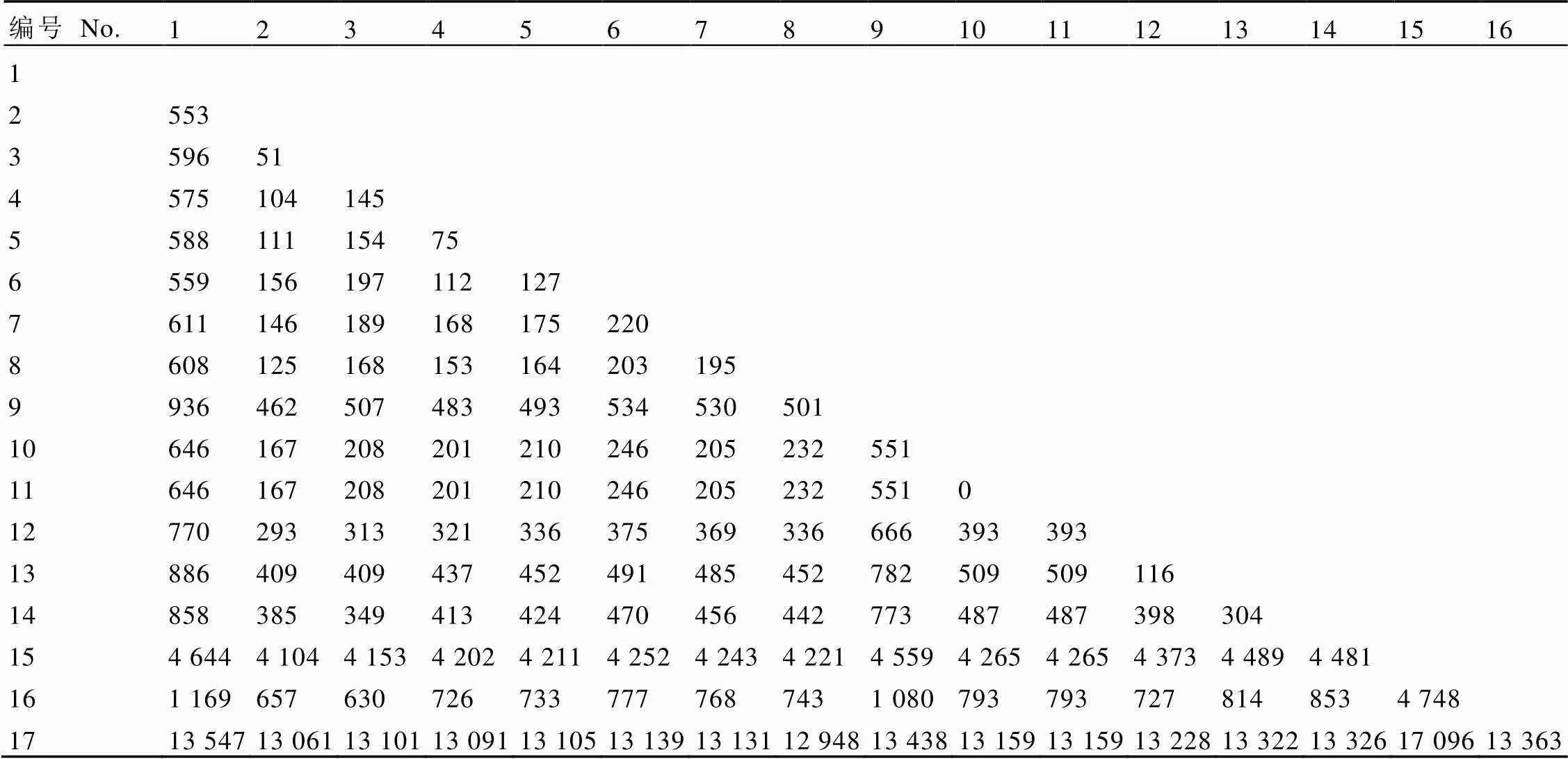
表5 17个茶树品种的叶绿体基因组碱基差异
注:编号1—17分别为白叶1号、白鸡冠、德宏茶、大红袍、肉桂茶、龙井43、安化茶、水金龟、武夷岩茶水仙、铁罗汉、信阳10号、云抗10号、大叶茶、白毛茶、sangmok、大叶茶(野生,MH394407)、铁观音
Note: Numbers 1-17 representcv. Baiye 1,cv. Baijiguan,var. Dehungensis,cv. Dahongpao,cv. Rougui,cv. Longjing 43,cv. Anhua,cv. Shuijinggui,cv. Wuyi Narcissus,cv. Tieluohan,cv. Xinyang 10,var. assamica cv. Yunkang 10,var. assamica,var. Pubilimba,cv. Sangmok,var. assamica (MH394407),cv. Tieguanyin, respectively
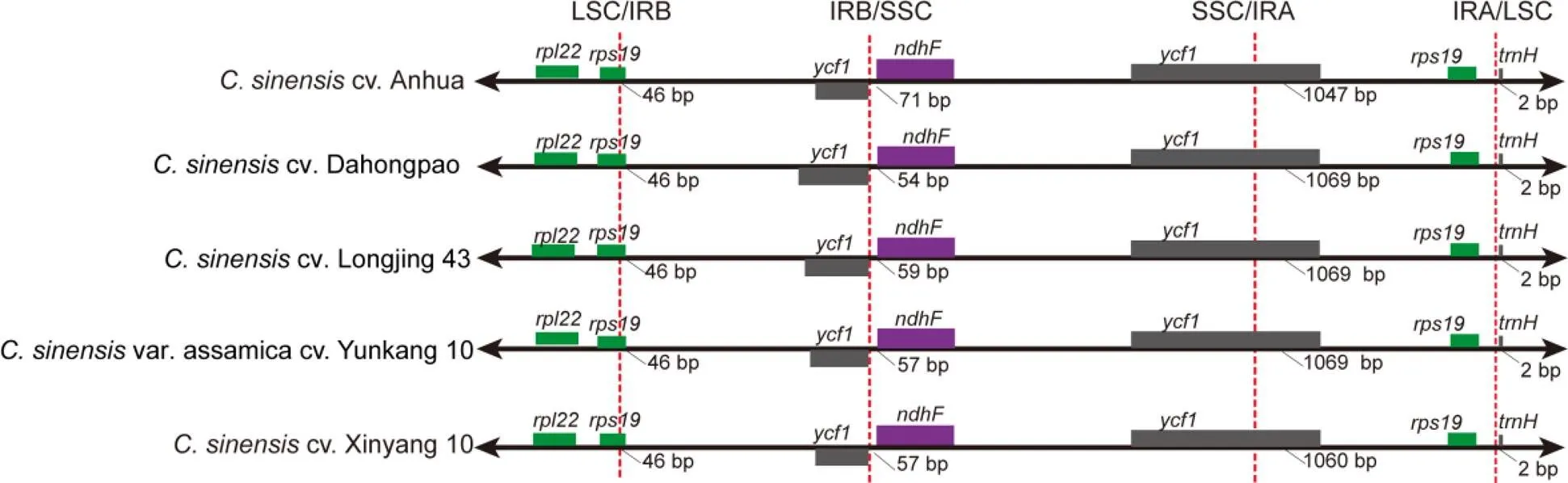
图2 5个茶树品种叶绿体基因组LSC、SSC和IR边界比较
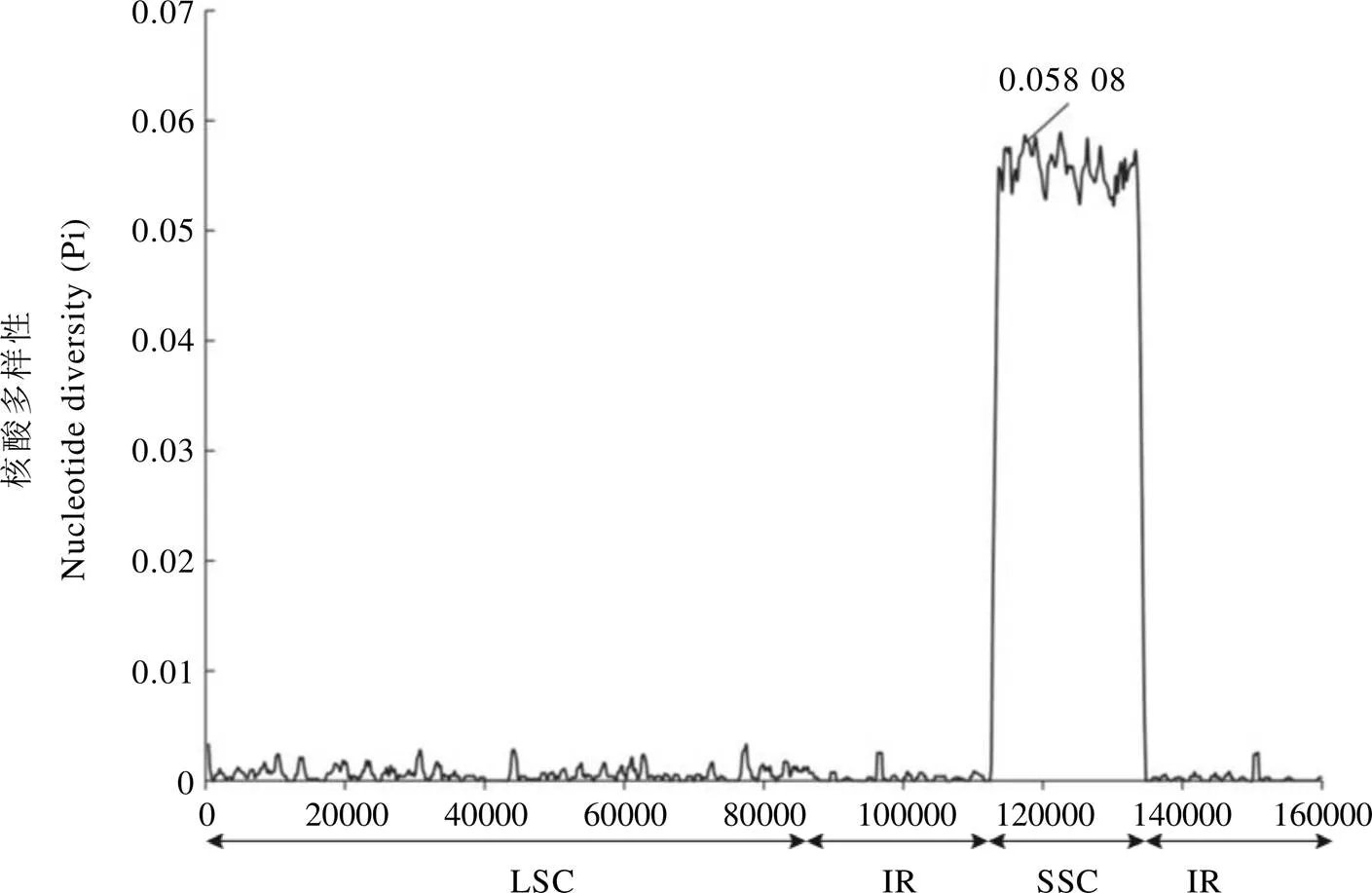
图3 17个茶树品种叶绿体基因组滑动窗口分析
2.4 系统进化分析
46个山茶属物种的贝叶斯进化分析显示,信阳10号与福建茶树品种铁罗汉聚为一支,支持率为100%,说明信阳10号与铁罗汉有较近的亲缘关系(图4)。云南德宏茶、膜叶茶(),福建白鸡冠以及两个韩国茶树品种形成进化树的内部分支,与信阳10号和铁罗汉分支形成姐妹群。浙江的龙井43与福建的肉桂茶和大红袍聚在一支,福建的水金龟、铁观音以及武夷岩茶水仙聚在一支。此外,选取的山茶属植物被大致分成了内部的茶亚属和外部的山茶亚属两个部分。然而并不是所有的茶亚属和山茶亚属的种都分别聚为一支,有一些茶亚属物种如和与山茶亚属的物种聚在一起,而山茶亚属的与茶亚属的种聚在一起,关系较近。
3 讨论
我国栽培茶树起源于西南地区,并向东和向北传播[26]。位于江北茶区的河南,年降水量较少,冬季寒冷,在此生长的茶树积累了适应本地气候的遗传信息,形成了独特的生理代谢,是茶树育种的宝贵资源。研究发现叶绿体基因参与了植物对环境的适应过程,如和基因与植物对热胁迫和干旱的适应有关[27];所有基因与光适应性调节有关[28]。此外,利用叶绿体基因组作为转基因的载体,不仅避免了通过花粉的转基因逃逸,还可以提高外源基因的表达[29]。本研究对信阳10号的叶绿体基因组进行测序和组装,不仅有助于探明信阳10号的亲本来源,还为研究茶树在中国中部的传播路径以及分子育种提供数据支持。
不同植物的叶绿体基因组提取方法存在差异,且较为复杂[30]。由于叶绿体基因组序列较为保守,通过全基因组测序,并以近缘物种的叶绿体基因组作为参考,从全基因组序列信息中提取叶绿体基因组成分的方法更为便捷[31]。本研究利用全基因组数据中分离出的叶绿体基因组reads,从头组装了国家级茶树良种信阳10号的叶绿体基因组,并对其结构进行了分析。
已测序并报道的茶树叶绿体基因组大小在153 044~157 353 bp[32-33],本研究组装的信阳10号的叶绿体基因组长度为157 041 bp,与福建铁罗汉叶绿体基因组长度相同,通过序列两两比较,发现铁罗汉与信阳10号的碱基组成也相同。地理来源方面,铁罗汉为福建品种,信阳10号来自于河南信阳本地茶树群体种,尽管两者相距较远,但根据叶绿体基因组完全相同以及叶绿体基因组母系遗传的特点,推断两者很可能来源于相同母本,并且分化时间不长。
叶绿体基因组的扩增和收缩是植物中常见的进化现象,其长度变化一般与IR区的扩张和收缩有关[34]。将信阳10号叶绿体基因组的IR区与湖南、浙江、云南和福建的茶树品种代表进行比较,其IR区长度属于中等水平,单个IR区长度为26 078 bp,小于云南云抗10号(26 083 bp)[10]和福建大红袍(26 079 bp)[11],大于湖南安化茶(26 068 bp)[13],与浙江龙井43相同[31]。5个茶树品种的叶绿体基因组GC含量基本相同,均在37.29%~37.30%,与已发表的大部分叶绿体基因组的GC含量相似[16]。
SSR广泛存在于基因组中,具有共显性[35]。SSR在叶绿体基因组中的拷贝数变异是一种重要的分子标记,在植物群体遗传学、多态性研究和系统进化研究中有着广泛的应用[36]。本研究经分析鉴定出信阳10号叶绿体基因组中的74个SSR,多分布于LSC区,主要由碱基A和T组成,这与山茶属叶绿体基因SSR分析结果类似[37]。SSR的类型在不同茶树品种中的分布也比较相似,不同茶树品种都以单核苷酸重复为主,其优势重复基元类型是A/T,信阳10号A/T型SSR占比与其他茶树相近,达到总数的75%[16,33]。SSR丰富的变异位点,将为今后开发SSR标记和进一步揭示茶树品种间进化关系提供有用的分子手段[38]。
利用46个叶绿体基因组构建的系统进化树揭示了信阳10号在不同茶树品种中的进化位置,发现福建的铁罗汉与信阳10号亲缘关系最近。利用叶绿体基因组构建的系统进化树在不同茶树品种间的分辨率较高,其中,17个茶树品种聚为一支,构成了进化树的内部类群,品种间的进化关系与以往的研究相似[39],本研究结果在不同品种形成的分支中混入了和,这与大红袍的叶绿体系统进化研究结果相同[11]。与以往山茶属分子系统学研究相比较,建树物种都被划分为山茶亚属和茶亚属[40]。然而两种类群并没有绝对分成两个部分,一些在张宏达系统[41]中被定为山茶亚属物种在进化树中与茶亚属聚在一起,茶亚属的部分物种也与山茶亚属聚在一起,说明山茶属的系统进化关系还有待进一步的解决。对更多的茶树叶绿体基因组进行测序,并将叶绿体基因组与核基因组信息共同分析或是解决茶树系统进化的有效途径。
信阳10号叶绿体全基因组序列与结构的揭示,为其遗传背景的研究和系统进化关系的探索奠定了基础。后续将以信阳10号叶绿体基因组作为参考序列,组装其他茶树栽培种和本地野生种的叶绿体基因组,对茶树的系统发育进行进一步探讨,同时结合环境因子,开展茶树本地适应性研究,为茶树分子育种提供遗传基础。
[1] Wei C L, Yan H, Wang S G, et al. Draft genome sequence ofvar.provides insights into the evolution of the tea genome and tea quality [J]. PNAS, 2018, 115(18): E4151-E4158.
[2] Chen L, Gao K Q, Chen M D, et al. The use of RAPD markers for detecting genetic diversity, relationship and molecular identification of Chinese elite tea genetic resources [(L.) O. Kuntze] preserved in a tea germplasm repository [J]. Biodiversity and Conservation, 2005, 14(6): 1433-1444.
[3] Chen L, Yamaguchi S. Genetic diversity and phylogeny of tea plant () and its related species and varieties in the sectiongenusdetermined by randomly amplified polymorphic DNA analysis [J]. Journal of Horticultural Science & Biotechnology, 2002, 77(6): 729-732.
[4] Vijayan K, Zhang W J, Tsou C H. Molecular taxonomy of(Theaceae) inferred from nrITS sequences [J]. American Journal of Botany, 2009, 96(7): 1348-1360.
[5] Yang H, Ling C L, Liu H W, et al. Genetic divergence betweenand its wild relatives revealed via genome-wide SNPs from RAD sequencing [J]. PLoS One, 2016, 11(3): e0151424. doi: 10.1371/journal.pone.0151424.
[6] Kaundun S S, Matsumto S. Molecular evidence for maternal inheritance of the chloroplast genome in tea,(L.) O. Kuntze [J]. Journal of the Science of Food and Agriculture, 2011, 91(14): 2660-2663.
[7] Huang H, Shi C, Liu Y, et al. Thirteenchloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships [J]. BMC Evolutionary Biology, 2014, 14: 151. doi: 10.1186/1471-2148-14-151.
[8] Alexander L W, Woeste K E. Pyrosequencing of the northern red oak (L.) chloroplast genome reveals high quality polymorphisms for population management [J]. Tree Genetics & Genomes, 2014, 10(4): 803-812.
[9] 张立, 程永琴, 姜在民, 等. 中国山茶科植物区系及叶绿体基因组结构进化分析[J]. 西北林学院学报, 2020, 35(5): 47-53.
Zhang L, Cheng Y Q, Jiang Z M, et al. Structure and phylogeny of chloroplast genomes and spermatophyte flora in Chinese Theaceae [J]. Journal of Northwest Forestry University, 2020, 35(5): 47-53.
[10] Gao L Z, Zhang F, Li W, et al. Deciphering tea tree chloroplast and mitochondrial genomes ofvar.[J]. Scientific Data, 2019, 6: 209. doi: 10.1038/s41597-019-0201-8.
[11] Li L, Hu Y F, Wu L H, et al. The complete chloroplast genome sequence ofcv.: a most famous variety of Wuyi tea (Synonym:L.) [J]. Mitochondrial DNA Part B, 2021, 6(1): 3-5.
[12] Hao W J, Wang S, Yao M, et al. The complete chloroplast genome of an albino tea,cultivar 'Baiye 1' [J]. Mitochondrial DNA Part B, 2019, 4(2): 3143-3144.
[13] Meng D, Liu S Q, Xu Z G, et al. The complete chloroplast genome of an economic plant,cultivar Anhua, China [J]. Mitochondrial DNA Part B, 2018, 3(2): 558-559.
[14] Prince L M, Parks C R. Phylogenetic relationships of Theaceae inferred from chloroplast DNA sequence data [J]. American Journal of Botany, 2001, 88(12): 2309-2320.
[15] Li W, Zhang C P, Guo X, et al. Complete chloroplast genome ofgenome structures, comparative and phylogenetic analysis [J]. PloS One, 2019. 14(5): e0216645. doi: 10.1371/journal.pone.0216645.
[16] Li L, Hu Y F, He M, et al. Comparative chloroplast genomes: insights into the evolution of the chloroplast genome ofand the phylogeny of[J]. BMC Genomics, 2021, 22(1): 138. doi: 10.21203/rs.3.rs-36917/v2.
[17] 张玲. 信阳毛尖产业化发展研究——以商城县为例[D]. 开封: 河南大学, 2015.
Zhang L. Research on the industrialization development of Xingyangmaojian: a case study of Shangcheng County [D]. Kaifeng: Henan University, 2015.
[18] 郭桂义. 中国名茶——信阳毛尖[J]. 信阳农业高等专科学校学报, 1999, 9(2): 49-52.
Guo G Y. Famous tea of China: Xinyangmaojian [J]. Journal of Xinyang Agricultural College, 1999, 9(2): 49-52.
[19] 朱德焰, 吕立哲, 金开美. 茶树良种信阳10号提纯复壮技术[J]. 广东农业科学, 2012, 39(2): 31-32.
Zhu D Y, Lv L Z, Jin K M. Purification and rejuvenation technology of tea variety Xinyang 10 [J]. Guangdong Agricultural Sciences, 2012, 39(2): 31-32.
[20] Langmead B, SalzbergS L. Fast gapped-read alignment with Bowtie 2 [J]. Nature Methods, 2012, 9(4): 357-359.
[21] Wick R R, Judd L M, Gorrie C L, et al. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads [J]. Plos Computational Biology, 2017, 13(6): e1005595. doi: 10.1371/journal.pcbi.1005595.
[22] Wyman S K, Jansen R K, Boore J L. Automatic annotation of organellar genomes with DOGMA [J]. Bioinformatics, 2004, 20(17): 3252-3255.
[23] Greiner S, Lehwark P, Bork R. Organellar Genome DRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes [J]. Nucleic Acids Research, 2019, 47(W1): W59-W64.
[24] Thiel T, Michalek W, Varshney R, et al. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (L.) [J]. Theoretical and Applied Genetics, 2003, 106(3): 411-422.
[25] Ronquist F, Huelsenbeck J P. MrBayes 3: bayesian phylogenetic inference under mixed models [J]. Bioinformatics, 2003, 19(12): 1572-1574.
[26] Xia E H, Tong W, Hou Y, et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation [J]. Molecular Plant, 2020, 13(7): 1013-1026.
[27] Caspermeyer J. Most comprehensive study to date reveals evolutionary history of[J]. Molecular Biology and Evolution, 2015, 32(8): 2217-2218.
[28] Peng L, Yamamoto H, Shikanai T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex [J]. Biochimica Et Biophysica Acta, 2011, 1807(8): 945-953.
[29] Jin S, Daniell H. The engineered chloroplast genome just got smarter [J]. Trends in Plant Science, 2015, 20(10): 622-640.
[30] 陈春梅, 陈亮. 茶树叶绿体DNA提取方法研究[J]. 分子植物育种, 2014, 12(3): 562-566.
Chen C M, Chen L. Extraction method of chloroplast DNA of tea plant () [J]. Molecular Plant Breeding, 2014, 12(3): 562-566.
[31] 叶晓倩, 赵忠辉, 朱全武, 等. 茶树“龙井43”叶绿体基因组测序及其系统进化(英文)[J]. 浙江大学学报(农业与生命科学版), 2014, 40(4): 404-412.
Ye X Q, Zhao Z H, Zhu Q W, et al. Chloroplast genome sequencing and phylogeny of tea plant Longjing 43 [J]. Journal of Zhejiang University (Agriculture and Life Sciences), 2014, 40(4): 404-412.
[32] Lee D J, Kim C K, Lee T H, et al. The complete chloroplast genome sequence of economical standard tea plant,L. cultivar Sangmok, in Korea [J]. Mitochondrial DNA Part B, 2020, 5(3): 2841-2842.
[33] Rawal H C, Borchetia S, Bera B, et al. Comparative analysis of chloroplast genomes indicated different origin for Indian tea (cv. TV1) as compared to Chinese tea [J]. Scientific Reports, 2021, 11(1): 110. doi: 10.1038/s41598-020-80431-w.
[34] Zhang X F, Landis J B, Wang H X, et al. Comparative analysis of chloroplast genome structure and molecular dating in Myrtales [J]. BMC Plant Biology, 2021, 21(1): 219. doi: 10.1186/s12870-021-02985-9.
[35] 李汝玉. 简单序列重复(SSR)及其在农作物研究中的应用[J]. 山东农业科学, 1999(4): 44-48.
Li R Y. Simple sequence repeat (SSR) and its application in crop research [J]. Shandong Agricultural Sciences, 1999(4): 44-48.
[36] 王化坤, 娄晓鸣, 章镇. 叶绿体微卫星在植物种质资源研究中的应用[J]. 分子植物育种, 2006, 4(3s): 92-98.
Wang H K, Lou X M, Zhang Z.Application in germplasm resource research using chloroplast simple sequence repeat [J]. Molecular Plant Breeding, 2006, 4(3s): 92-98.
[37] Zhang W, Zhao Y L, Yang G Y, et al. Determination of the evolutionary pressure onon Hainan Island using the complete chloroplast genome sequence [J]. Peerj, 2019, 7: e7210. doi: 10.7717/peerj.7210.
[38] Liu S R, An Y L, Li F D, et al. Genome-wide identification of simple sequence repeats and development of polymorphic SSR markers for genetic studies in tea plant () [J]. Molecular Breeding, 2018, 38: 59. doi: 10.1007/s11032-018-0824-z.
[39] Chen S, Li R Y, Ma Y Y, et al. The complete chloroplast genome sequence ofvar.cultivar Tieguanyin (Theaceae) [J]. Mitochondrial DNA Part B, 2021, 6(2): 395-396.
[40] 江正栋. 基于叶绿体DNA的山茶属植物的分子系统学和生物地理学初探[D]. 杭州: 浙江理工大学, 2017.
Jiang Z D. Preliminary study of molecular phylogenetics and biogeography of the genusL. based on chloroplast DNA [D]. Hangzhou: Zhejiang Sci-Tech University, 2017.
[41] 张宏达, 任善湘. 中国植物志: 第四十四卷第一分册[M]. 北京: 科学出版社, 1998: 48-91.
Zhang H D, Ren S X. Flora of China: Vol.44, Part 1 [M]. Beijing: Science Press, 1998: 48-91.
Complete Chloroplast Genome ofcv. Xinyang 10 and Its Phylogenetic Evolution
YAN Minghui1, LIU Ke2, WANG Man2, LYU Ying1, ZHANG Qian1
1. College of Life Science, Xinyang Normal University/Henan Key Laboratory of Tea Plant Biology, Xinyang 464000, China; 2. College of International Education, Xinyang Normal University, Xinyang 464000, China
cv. Xinyang 10 is a national excellent cultivar suitable for producing Xinyangmaojian. However, its origin and evolutionary relationship with other tea cultivars are still unknown. This study obtained the complete chloroplast genome ofcv. Xinyang 10 by using MGI2000 sequencing platform, and then analyzed the chloroplast genome structure. A chloroplast genome phylogenetic analysis of 46 species was also conducted to infer the position ofcv. Xinyang 10. The results show that: (1) the chloroplast genome of Xinyang 10 is 157 041 bp in length, including a pair of inverted repeat regions (IR, 26 078 bp), a large single-copy region (LSC, 86 594 bp) and a small single-copy region (SSC, 18 291 bp). A total of 113 genes are annotated, including 79 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. (2) 74 SSR loci were detected in the chloroplast genome of Xinyang 10, most of the SSRs were composed of A/T. (3) The cluster analysis using Bayesian method shows that Xinyang 10 had the closest genetic relationship withcv. Tieluohan. Both cultivars may share the same female parent as their chloroplast genomes were identical. Xinyang 10 also had a close relationship with two Korean tea (Chamnok and Sangmok),Baijiguan andvar. Dehungensis. The results provided a basis for further research on the origin and evolution of tea and molecular breeding.
,cv. Xinyang 10, chloroplast genome, phylogentic analysis
S571.1
A
1000-369X(2021)06-777-12
2021-06-16
2021-07-26
国家自然科学基金(31800276)、河南省高等学校重点科研项目(19A180029)、信阳师范学院“南湖学者奖励计划”青年项目
闫明慧,女,讲师,主要从事植物系统进化研究,yanminghui8899@163.com
(责任编辑:黄晨)
猜你喜欢
人大建设(2020年5期)2020-09-25 08:56:18
文苑(2020年6期)2020-06-22 08:41:50
青年歌声(2018年8期)2018-10-22 01:16:20
青年歌声(2018年2期)2018-10-20 02:02:56
大观(书画家)(2018年6期)2018-07-08 00:43:20
民族音乐(2016年1期)2016-08-28 20:02:52
电影故事(2016年5期)2016-06-15 20:27:30
广西林业科学(2016年1期)2016-03-20 05:33:01
小溪流(画刊)(2014年6期)2014-08-11 19:25:34
食品科学(2013年6期)2013-03-11 18:20:13
