
铁死亡与阿尔茨海默病的研究进展*
2021-12-06 05:16:58安红伟曹诗杰
中国病理生理杂志 2021年11期
黄 健, 安红伟, 曹诗杰
(1广西中医药大学,广西南宁530001;2柳州市中医医院,广西柳州545001)
阿尔茨海默病(Alzheimer disease,AD)常表现为进行性认知功能障碍与行为损害,也是痴呆最常见的病因,目前全世界约有5 000 万患者。AD 的病理特征是β-淀粉样蛋白(amyloid β-protein,Aβ)沉积形成细胞外老年斑(senile plaques,SPs)和高磷酸化tau 蛋白形成细胞内神经原纤维缠结(neurofibrillary tangles,NFTs)。此外,AD 患者神经元进行性死亡,伴有氧化应激和铁超载。Dixon 等[1]于 2012 年首次报道铁死亡(ferroptosis)是一种以细胞内铁沉积为特征的细胞死亡形式。铁沉积诱导氧化应激产生活性氧(reactive oxygen species,ROS)并导致脂质过氧化物(lipid peroxide,Lipid-OOH)的积累[2]。已有研究观察到AD 患者脑细胞表现出类似铁死亡的生化和形态学特征,包括谷胱甘肽(glutathione,GSH)降解、谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)失活、铁代谢失衡导致ROS 增加、脂质过氧化及线粒体异常[3],并且铁代谢紊乱与Aβ、SPs 和NFTs密切相关。本文拟对铁死亡在AD 中的作用及机制进行综述。
1 脑细胞的铁代谢
1.1 脑细胞的铁转运机制
1.1.1 铁流入分子机制 细胞转运铁的方式有转铁蛋白(transferrin,Tf)和非转铁蛋白两种。生理条件下Fe3+通过管腔毛细血管内皮细胞上的转铁蛋白受体(transferrin receptor,TfR)与Tf 介导内吞作用而被转入细胞。尽管Tf 与铁连接的饱和度通常为30%[4],但疾病导致组织细胞铁超载,Tf 与铁连接饱和,多余的血浆铁以非Tf铁的形式出现。Fe3+被核内体(endosome)金属还原酶(metalloreductase)前列腺6次跨膜上皮抗原3(six-transmembrane epithelial antigen of the prostate 3,Steap3)还原为Fe2+,然后可由二价金属转运蛋白1(divalent metal transporter 1,DMT1)或锌转运蛋白 ZIP14(ZRT/IRT-like protein 14)协助而流入胞质中的不稳定铁池(labile iron pool,LIP)[5]。
1.1.2 铁流出分子机制 膜铁转运蛋白1(ferropor-tin 1,FPN1)是哺乳动物细胞内仅有转运铁至胞外的蛋白[6]。在神经元中,淀粉样前体蛋白(amyloid precursor protein,APP)与 FPN1 连接并稳定 FPN1 的表达水平,同时APP 被认为是一种铁氧化酶,可将Fe2+氧化为 Fe3+并转出细胞[7]。铁调素(hepcidin)主要由肝细胞合成和分泌,它与FPN1结合并触发其内化、泛素化和降解[8]。AD 大鼠模型和患者脑中APP水平被下调,同时皮层和海马神经元FPN1基因敲除导致类似AD的海马萎缩和认知功能下降[9]。
1.1.3 铁在脑细胞的吸收 不同的细胞类型有不同的铁吸收方式,神经元和小胶质细胞可通过Tf/TfR1 途径获取铁,或通过DMT1 依赖的管腔途径吸收非 Tf 结合铁(non-transferrin-bound iron,NTBI)[10]。最近的研究表明,H-铁蛋白(H-ferritin)与H-铁蛋白受体的结合可能是少突胶质细胞铁摄取的主要来源[11]。除了 Tf/TfR1 或 DMT1 通路外,星形胶质细胞还可能通过其末端足突吸收铁[12]。铁经上述不同方式通过血脑屏障(blood-brain barrier,BBB)被神经元、小胶质细胞和星形胶质细胞吸收。
1.2 脑细胞铁代谢调节机制 脑细胞铁代谢失衡——铁超载或铁储备不足与AD 有关。铁蛋白与FPN 协同工作,在铁氧化酶(ferroxidase)和铜蓝蛋白(ceruloplasmin)帮助下调节LIP。LIP 调控铁调节蛋白1(iron regulatory protein 1,IRP1)和IRP2 的激活,而IRP1和IRP2又与mRNA 铁调节序列[称为铁反应元件(iron response elements,IREs)]结合。不稳定铁以Fe3+复合物的形式储存在铁蛋白中,并通过溶酶体(lysosome)或自噬体(autophagosome)降解释放。LIP内增加的铁导致铁超载,激活Fenton 和Haber-Weiss反应,从而产生自由基,损伤蛋白质和脂质等。通过Fenton反应,Fe2+被H2O2氧化为Fe3+,形成高活性的羟基自由基(·OH);通过Haber-Weiss 反应,超氧化物歧化作用将Fe3+转化为Fe2+,产生超氧自由基(·O2-),诱导氧化应激[13]。AD 患者脑内铁蛋白重链和轻链表达增加,提示 LIP 扩增[14]。Ayton 等[15]对 209 例 AD患者连续12 年随访研究的结果显示,颞下皮质铁含量升高,提示铁与认知能力下降密切联系。铁代谢失调与AD 病理改变密切相关,脑铁水平与AD 病程进展和认知下降呈正相关[16]。见图1。
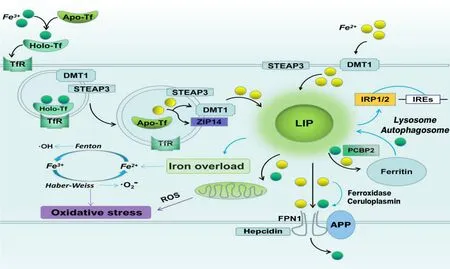
Figure 1. The mechanism of iron metabolism. The black arrow represents the molecular mechanism of iron transport. The blue arrow represents the regulation mechanism of iron metabolism homeostasis. The purple arrow represents the process of oxidative stress.图1 铁代谢的机制
2 铁死亡
2.1 铁死亡简介 铁死亡是由于铁代谢失衡,ROS堆积导致脂质过氧化进而损伤细胞,GSH 减少和GPX4 失活常诱导铁死亡。电子显微镜下铁死亡细胞表现出线粒体变小、线粒体膜皱褶、线粒体嵴减少和外膜破裂,而线粒体在其他形式的细胞死亡中通常表现肿胀。ferrostatin-1、liproxstatin-1 和维生素E抵抗铁死亡诱导的细胞死亡,但上述抑制剂不能抑制其他形式的细胞死亡。在铁死亡中,铁蛋白的自噬即铁自噬(ferritinophagy)使得LIP 的增加,导致Lipid-OOH 积累和多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)氧化。通过Ras 选择性致死小分子 3(Ras-selective lethal small molecule 3,RSL3)抑制GPX4,或敲除GPX4基因可诱导铁死亡。铁死亡抑制蛋白1(ferroptosis suppressor protein 1,FSP1)通过清除GPX4 介导的NAD(P)H 还原辅酶Q10,抑制脂质过氧化,从而发挥抗铁死亡作用。
2.2 铁代谢参与铁死亡 铁代谢平衡依赖多种铁代谢相关蛋白的协同作用,铁代谢紊乱可能引发铁死亡。Tf/TfR1 通路主要负责细胞吸收铁,当Tf/TfR1通路转运铁功能异常会导致氧化应激和细胞铁死亡。TfR1循环和棕榈酰化异常可导致脑铁沉积神经变性病变[17]。研究表明作为吸收铁的蛋白DMT1 过表达会导致黑质铁沉积和多巴胺能神经元死亡[18]。铁蛋白是细胞内由轻链蛋白和重链蛋白组成的主要存储铁的蛋白,以氧化还原态非活性形式储备铁,并防止细胞和组织受到氧化损伤。铁蛋白异常引起的铁代谢失衡将诱发铁死亡[19]。铁死亡时胞质内铁蛋白会在溶酶体内发生铁自噬,进一步扩大LIP,铁自噬增强半胱氨酸缺乏诱导的铁死亡。虽然细胞内总铁含量可能没有变化,但LIP扩大使细胞容易发生铁死亡。铁代谢失衡导致铁超载,再通过Fenton 和Haber-Weiss反应产生自由基,从而激活铁死亡。
2.3 GSH/GPX4信号通路通过介导脂质过氧化参与铁死亡 膜脂中的PUFAs,特别是花生四烯酸(arachidonic acid,AA),在铁死亡时优先被脂氧合酶(lipoxygenases,LOXs)氧化而导致细胞损伤。AA 被酰基辅酶A 合成酶长链家族成员4(acyl-CoA synthetase long-chain family member 4,ACSL4)激活,并入磷脂酰乙醇胺(和膜),而溶血卵磷脂酰基转移酶3(lysophosphatidylcholine acyltransferase 3,LPCAT3)对 AA 并 入 膜 磷 脂 很 重 要[20]。 失 活 的 ACSL4 或LPCAT3 抑制铁死亡比抑制其他形式的细胞死亡效果要强。参与脂质过氧化的LOXs 在其活性部位需要铁来催化,铁螯合剂[去铁胺(deferoxamin)和去铁酮(deferiprone)]通过去除LOXs 中必需的铁来抑制铁死亡[21]。花生四烯酸 15-脂氧合酶(arachidonate 15-lipoxygenase,ALOX15)通过促进Lipid-OOH 的形成在erastin诱导的铁死亡中发挥重要作用[20]。
神经元特异性GPX4基因敲除已被认为会导致神经元死亡,GPX4的硒代半胱氨酸残基突变激活铁死亡[22]。GPX4 将 GSH 作为底物介导 Lipid-OOH 转换为Lipid-OH。GSSG 在NADPH 参加的反应中被谷胱甘肽还原酶还原为GSH。GSH 是由谷氨酸-半胱氨酸连接酶(glutamate-cysteine ligase,GCL)和谷胱甘肽合成酶(glutathione synthetase,GS)连续作用合成的,其中GCL 是GSH 合成的限速步骤。半胱氨酸通过半胱氨酸/谷氨酸逆向转运体(system XC-)交换谷氨酸。通过抑制system XC-减少半胱氨酸的供应,减弱GSH 水平,可使GPX4 失活。当GPX4 催化的脂质反应不足以抑制铁介导的脂质自由基生成Lipid-OOH时,将诱发铁死亡[23]。
3 铁死亡在AD中的作用机制
3.1 GSH/GPX4 参与的铁死亡在AD 中作用及机制 研究表明,海马和额叶皮质的GSH 水平减少与严重的认知障碍有关[15],提示GSH有可能成为AD的生物标志物。通过口服GSH 补充剂恢复大脑中的GSH 水平效果不佳,因为GSH 会迅速水解并且血脑屏障渗透不足。同样,L-半胱氨酸(GSH 合成的限速底物)由于广泛代谢而不足。然而,作为L-半胱氨酸前体的N-乙酰半胱氨酸(N-acetyl-L-cysteine,NAC)可以有效地穿过血脑屏障进入大脑,在AD 大鼠模型中NAC 调控大脑GSH 水平而发挥抗脂质过氧化作用,使用NAC可增强细胞膜和线粒体膜的通透性,从而增加GSH 水平,抑制铁死亡,发挥神经保护作用[24-25]。前脑神经元GPX4 失活诱发海马神经元死亡,具有特定大脑皮层和海马神经元GPX4基因敲除小鼠在水迷宫试验中表现出明显的认知障碍及海马神经元退行性改变;硫辛酸(thioctic acid)通过下调TfR 和上调GPX4 表达来抑制铁死亡,从而挽救学习和记忆相关的神经元[23,26]。
3.2 铁死亡与Aβ和APP 相互作用 Aβ在将Fe3+还原为Fe2+时导致过量自由基生成并诱导神经元损伤。APP 是 1 型跨膜糖蛋白,是产生 Aβ 的前体。APP 首先被α-分泌酶或β-分泌酶分解,然后被γ-分泌酶分解。生理状态下α-分泌酶首先切割APP进行非淀粉样变途径。然而,若APP 首先被β-分泌酶切割会产生具有神经毒性的Aβ。而furin 蛋白在介导α-分泌酶或β-分泌酶的蛋白水解激活率方面起着关键作用,furin蛋白的浓度与α-分泌酶活性呈正相关,与β-分泌酶活性呈负相关。铁超载导致furin转录翻译减少。铁通过抑制furin 蛋白表达增强β-分泌酶活性,从而使通过淀粉样变途径产生的Aβ 增加[27]。已有研究证实,受损APP 的N-糖基化或磷酸化阻碍APP向细胞膜的转运,从而干扰FPN1 的稳定性,降低细胞膜FPN1 的水平,铁转运能力显著降低,导致细胞内铁超载,从而诱导铁死亡[28]。轻度认知障碍伴多发Aβ 斑块的患者皮质铁含量较高,从而增加AD风险。
3.3 铁死亡与tau 蛋白和NFTs 相互作用 脑铁稳态失调与tau蛋白和NFTs密切相关。生理条件下tau蛋白是一种促进神经元微管结构形成的相关蛋白,在轴突运输和认知功能中发挥关键作用。然而,tau蛋白过度磷酸化聚集成NFTs。铁可以调节tau 蛋白磷酸化并诱导高磷酸化tau 蛋白的聚集。研究表明,tau 蛋白通过稳定神经元微管在铁转运中起重要作用,在正常生理学中,tau 蛋白可以通过转运至细胞膜来调节细胞铁外流,tau 蛋白的过度磷酸化和聚集还可能影响铁外流,导致神经元铁沉积和加重NFTs,引起恶性循环[29]。此外铁沉积与神经退行性变区域NFTs存在共定位现象[30]。
4 调控铁死亡对AD的作用
自由基清除剂、铁螯合剂和GPX4类似物等能够抑制铁死亡,从而防治AD。研究表明去铁酮和NAC联合治疗AD 比单一治疗更能对抗铁诱导的毒性损伤,从而保护神经,包括恢复树突棘密度和线粒体动力学稳态[31]。
4.1 调控铁死亡中GPX4对AD的作用 GPX4是铁死亡的关键因子。RSL3、ML-162 和ML-210 与GPX4结合而抑制铁死亡,FIN56 则通过抑制GPX4 活性而诱导铁死亡。研究者在HT22 神经细胞内发现RSL3抑制GPX4活性,导致线粒体损伤和脂质过氧化。α-生育酚、脂氧合酶抑制剂或沉默凋亡诱导因子(apoptosis-inducing factor,AIF)能够阻止GPX4 失活。Ebselen 是一种脂溶性抗氧化剂,具有类似GPX 活性。Ebselen 处理后AD 模型小鼠的空间和记忆功能明显改善,Aβ、p-tau 蛋白和突触后密度蛋白水平降低,并且海马神经元死亡减少[32]。AD 模型中海马、颞叶和皮层区域硒含量降低,与氧化应激增强一致。硒被神经元用来合成作为硒代半胱氨酸载体的GPX4。AD 患者血液铁离子水平升高与硒含量下降和认知障碍相关,提示补充硒是一种潜在干预铁死亡和神经退行性变方法[33]。
4.2 自由基清除剂调控铁死亡对AD 的作用 过量的游离铁(Fe3+/Fe2+)因具有氧化还原性能产生大量自由基而激发脂质过氧化。自由基清除剂(如ferrostatin-1和liproxstatin-1)干扰脂质自由基氧化。ferrostatin-1是一种通过阻断ROS产生和脂质过氧化的铁死亡抑制剂,并且能抑制谷氨酸诱导的大鼠脑细胞死亡。ferrostatin-1 和liproxstatin-1 还能抑制erastin诱导的小鼠海马神经元线粒体功能障碍和细胞死亡[34]。维生素E 可以被认为是铁死亡的抑制因素,AD 患者血浆、血清和脑脊液中的维生素E 减少,补充维生素E 可延缓轻度至中度AD 患者的功能下降[35]。维生素E 中和氧自由基,从而终止脂质(特别是PUFAs)的过氧化。中年人长期补充PUFAs 可保护细胞膜免受脂质过氧化,降低症状前期的AD 风险[36]。给予大鼠高维生素E 饮食或给予铁死亡抑制剂liproxstatin-1时,神经元退行性改变水平降低[37]。
4.3 铁离子螯合剂调控铁死亡对AD 的作用 铁螯合剂可抑制铁死亡,从而恢复细胞功能。低亲和力铁螯合剂调控铁代谢紊乱,抑制细胞铁死亡,进而治疗AD。去铁胺可减轻AD 患者的认知能力下降,然而,由于缺乏去铁胺穿透BBB 的研究,去铁胺治疗没有进一步研究。铁螯合作用减弱氧化应激,降低Aβ水平和tau 蛋白过度磷酸化。患者接受铁螯合剂治疗最严重的不良反应是中性粒细胞减少(8.5%)[38],因此定期评估铁代谢的主要器官肝肾的功能至关重要。
4.4 system XC-抑制剂调控铁死亡对AD的作用 抑制system XC-使细胞缺乏半胱氨酸,从而使GSH 水平降低,GPX4 失活。半胱氨酸供应可被system X-抑
C制剂[包括erastin、柳氮磺胺吡啶(sulfasalazine)和索拉非尼(sorafenib)]阻断。erastin 通过ALOX15 促进Lipid-OOH 的形成,从而诱导铁死亡[20]。而激活system XC-使谷氨酸转出增加,可激活抑制性代谢型谷氨酸受体,从而减轻丘脑皮层由谷氨酸过多引起的认知损害,这种保护作用可能与抑制细胞铁死亡有关[39]。
铁死亡与AD的关系总结见图2。
5 总结与展望
AD 患者脑细胞中铁超载引发氧化应激损伤,加重毒性 Aβ 和过度磷酸化 tau 蛋白聚集[40]。Huang等[41]的研究显示,Aβ 使细胞产生氧化应激或诱导铁死亡,但是Aβ 与铁死亡关系仍需深入研究。铁死亡中脂质过氧化与氧化应激关系密切并且研究较多,但铁死亡与其他形式的细胞死亡诸如自噬、凋亡和胀亡之间的关系需要更深入研究。目前讨论铁主要集中在LIP,但体内95%的功能性铁是血红素,因此,需要进一步的研究来验证血红素的铁在AD 中的作用。Gleason 等[42]的研究提示,AD 病变区域铁超载可能是认知能力迅速下降的危险因素。当前许多旨在单纯降低Aβ 或tau 蛋白来治疗AD 的临床试验没有成功,可能是部分AD患者表现出α-突触核蛋白和TDP43 改 变。 2021 年 6 月美 国 FDA 加 速批 准 针对Aβ 的创新药Aducanumab 上市引起争议。因此,需要深入探讨铁死亡在AD 中的作用及机制,以期为AD患者提供治疗新靶点。

Figure 2. The relationship between ferroptosis and Alzheimer disease(AD). The red arrow represents the changes in the key factors of ferroptosis in AD. The black arrow represents the mechanism of GSH/GPX4 pathway in ferroptosis. The cyan arrow represents the molecular mechanism of lipid peroxidation. The blue arrow represents the activation/inhibition mechanism of ferroptosis.图2 铁死亡与阿尔茨海默病的关系
猜你喜欢
科学(2022年4期)2022-10-25 02:43:32
现代临床医学(2021年6期)2021-11-20 06:34:50
中成药(2017年12期)2018-01-19 02:06:48
现代检验医学杂志(2016年5期)2016-08-20 03:17:14
天津科技大学学报(2015年3期)2015-04-16 04:54:59
中国当代医药(2015年22期)2015-03-01 02:05:16
中国洗涤用品工业(2015年4期)2015-02-28 19:02:15
安徽医专学报(2014年6期)2014-03-20 13:08:05
食品工业科技(2014年15期)2014-03-11 18:17:43
中国神经精神疾病杂志(2013年1期)2013-03-11 20:23:37
