
代谢重编程在肺动脉高压发病机制中的研究进展*
2021-12-06 05:16:58齐先梅
中国病理生理杂志 2021年11期
齐先梅, 罗 娅, 王 婧
(中国医学科学院基础医学研究所,北京协和医学院基础学院,北京100730)
肺动脉高压(pulmonary hypertension,PH)是一组严重的进行性心肺疾病,肺血管收缩、肺中小动脉重构及原位血栓形成引起肺血管阻力逐渐增加,最终导致右心衰竭甚至死亡[1-2]。目前认为促进PH 发生发展的关键机制是血管细胞的过度增殖和/或抗凋亡[3],同时血管收缩、炎症及纤维化也参与其中[4-5]。PH 患病人数庞大,发病率逐年升高[6],由于对PH 发生的病理生理机制认识不全,目前尚无治愈方法[7]。代谢重编程通过供给能量、生物合成中间物和还原性物质,维持细胞的氧化还原稳态并保证生物分子合成,使细胞呈现增殖、迁移以及抗凋亡的肿瘤样恶性表型,导致肺血管重构和右心室肥厚,加速PH 的发生发展[7]。此外,由于代谢可调节与PH发生发展有关的多种因素(如遗传、低氧、氧化应激、炎症等)[8-9],提示代谢重编程可能是PH 疾病过程中的关键环节。因此,从代谢角度研究PH 的病理生理机制对探索潜在的靶向药物具有重要意义。本综述将着重对PH 中糖、脂肪酸和氨基酸代谢重编程的研究进展进行总结。
1 PH中的糖代谢重编程
葡萄糖分解代谢主要包括3 条途径:糖酵解(又称糖无氧氧化)途径、糖有氧氧化途径和磷酸戊糖途径(pentose phosphate pathway,PPP),代谢途径简图见图1。有氧糖酵解或称Warburg 效应是在肿瘤细胞中发现的一种糖酵解的特殊形式,指在氧气充足的条件下,细胞仍偏好糖酵解[10],可使葡萄糖摄取量大幅度增加,满足快速增殖细胞的能量需求,并为生物大分子合成提供物质基础,促进细胞的生长、增殖、转移等肿瘤样恶性表型形成[3,11]。目前,PH 中葡萄糖氧化减少,而Warburg效应和PPP途径增强已被广泛报道。
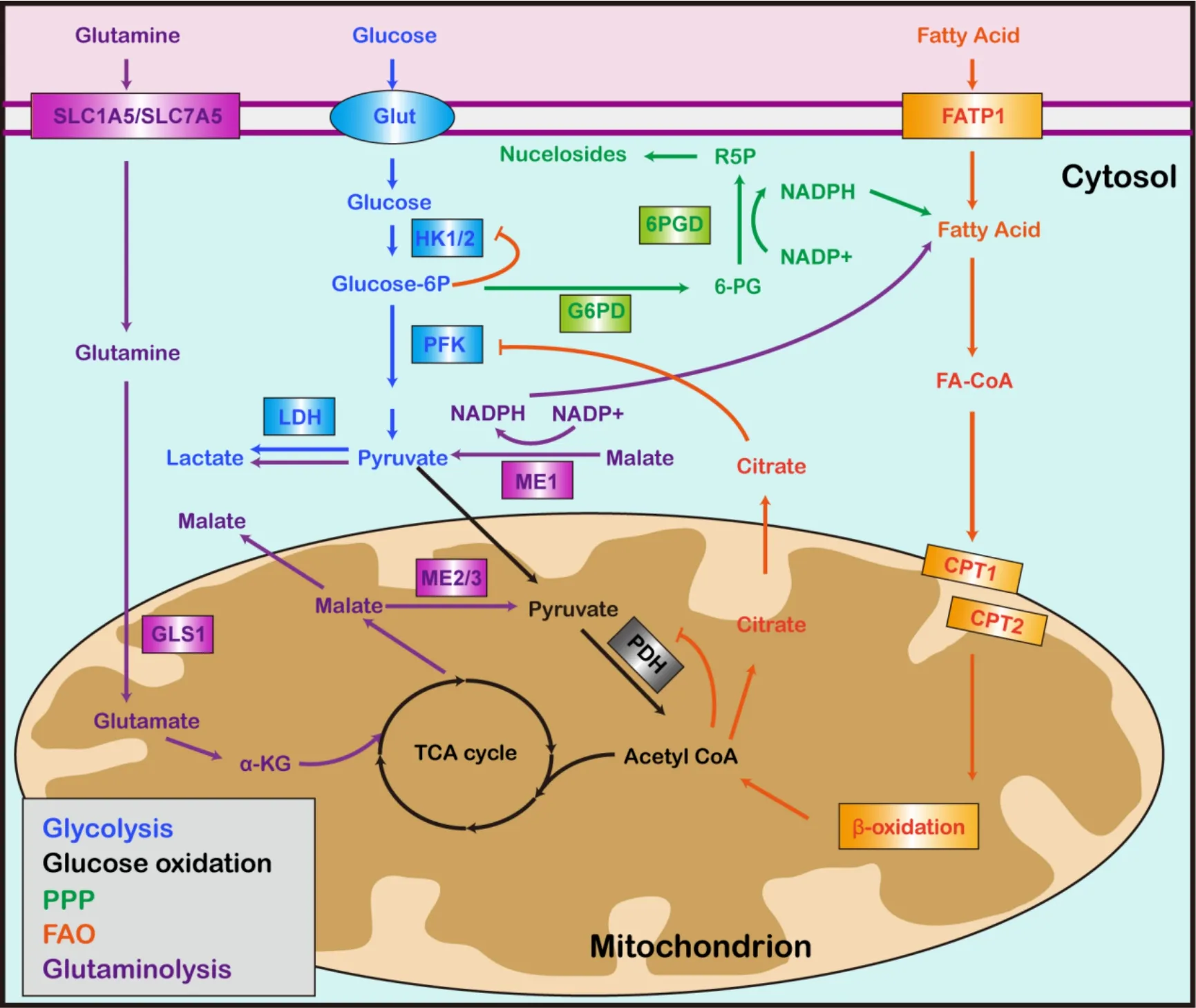
Figure 1. Schematic illustration of metabolic pathways. PPP:pentose phosphate pathway;FAO:fatty acid oxidation;Glut:glucose transporter;HK1/2:hexokinase 1/2;PFK:phosphofructokinase;LDH:lactate dehydrogenase;PDH:pyruvate dehydrogenase;TCA:tricarboxylic acid;G6PD:glucose 6 phosphate dehydrogenase;6PGD:6-phosphogluconate dehydrogenase;6-PG:6-phosphogluconate;NADPH:nicotinamide adenine dinucleotide phosphate;R5P:ribose-5-phosphate;FATP1:fatty acid transport protein 1;FA-CoA:fatty acyl-CoA;CPT1/2:cholinephosphotransferase 1/2;SLC1A5:solute carrier family 1 member 5;SLC7A5:solute carrier family 7 member 5;GLS1:glutaminase-1;ME:malic enzyme;α-KG:α-ketoglutarate.图1 代谢途径简图
1.1 Warburg 效应 PH 疾病过程中,糖代谢相关酶,包括葡萄糖转运体、己糖激酶2、α-烯醇化酶、丙酮酸脱氢酶激酶1(pyruvate dehydrogenase kinase 1,PDK1)、乳酸脱氢酶等表达增加或丙酮酸脱氢酶(pyruvate dehydrogenase,PDH)被抑制,将导致Warburg效应出现[3,12-14],促进肺血管细胞,包括内皮细胞(endothelial cells,ECs)、平滑肌细胞(smooth muscle cells,SMCs)和成纤维细胞(fibroblasts,Fibs),及右室心肌细胞的肿瘤样恶性表型形成[4,11,13-14],导致肺动脉收缩及重构、右心室收缩力降低和心输出量减少,促进PH的发生发展。
Warburg效应促进PH 中关键细胞形成肿瘤样恶性表型的具体机制目前尚不十分清楚,但在PH 中Warburg 效应的调节途径已得到部分认识。糖酵解途径可受表观遗传学改变、基因突变以及其他多种通路的调控[15]。根据以往研究,可将Warburg效应的调节途径归纳为3类(图2)。对于Warburg调节途径1,研究表明DNA 甲基化参与了PH 的发生发展,当DNA 甲基转移酶(DNA methyltransferase,DNMT)1和DNMT3b的表达增加时,超氧化物歧化酶2活性下降有助于低氧诱导因子1α 激活[16],从而抑制PDK 的活性,导致Warburg 效应,促进肺血管细胞过度增殖和形成抗凋亡表型。目前,该调节通路已在肺动脉平滑肌细胞(pulmonary arterial smooth muscle cells,PASMCs)和右室心肌细胞中被证实。二氯乙酸盐可抑制该途径,减轻血管重塑和部分恢复右心室收缩性,从而缓解 PH[17-18]。对于 Warburg 调节途径 2,相关研究显示microRNA-124 下调可促进肺动脉内皮细胞(pulmonary arterial endothelial cells,PAECs)、SMCs 和Fibs 的RNA 剪接因子聚嘧啶束结合蛋白1(polypyrimidine tract-binding protein 1,PTBP1)上调[12,19-20]。PTBP1 可调节丙酮酸激酶(pyruvate kinase,PK)亚型的剪接,当PTBP1 增加时可导致M2型 PK(PKM2)活性增加(PAECs 和 PASMCs)或者PKM2/PKM1 比值增加(Fibs),增强Warburg 效应,促进肺血管细胞过度增殖和形成抗凋亡特性,从而加重PH 的发生发展。同时,通过药理学方法抑制PKM2 活性/表达或者靶向抑制microRNA-124 表达,使PKM2/PKM1 比值恢复正常,可在体外和体内实验中抑制肺血管细胞过度增殖及 PH 进展[12,19]。此外,其他调节方式包括活化T细胞核因子[21]、C 端结合蛋白 1[13]、6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶 3[22]、CD146[23]、microRNA-25、microRNA-138[24]等均可促进Warburg效应。

Figure 2. Regulatory pathways of Warburg effect in PH[4]. Glut:glucose transporter;HK1/2:hexokinase 1/2;PEP:phosphoenolpyruvate;PKM:pyruvate kinase M;LDH:lactate dehydrogenase;PDH:pyruvate dehydrogenase;PDK:pyruvate dehydrogenase kinase 1;TCA:tricarboxylic acid;DNMT:DNA methyltransferase;SOD2:superoxide dismutase 2;HIF-1α:hypoxia-inducible factor 1α;BMPR2:bone morphogenetic protein receptor type-2;miRNA:microRNA;PTBP1:polypyrimidine tract-binding protein 1;HRE:hypoxicresponseelements;CtBP1:transcriptional corepressor c-terminal binding protein-1;PFKFB3:6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3;MCUC:mitochondrial calcium uniporter complex.图2 PH中Warburg效应的调节途径[4]
尽管在PAECs、SMCs、Fibs及右室心肌细胞中的关键酶/调节信号通路可能存在一定差异,但增强的Warburg 效应可通过促进这些细胞出现过度增殖和抗凋亡表型,促进肺血管重塑和右室肥厚。而通过药理学方法抑制或表观遗传调控PH中的Warburg效应,可缓解PH 的进展,因此糖酵解途径的关键酶可能是PH潜在的治疗靶点。
1.2 PPP 该途径分为两个阶段。在第一阶段的氧化反应中,葡萄糖-6-磷酸在葡萄糖-6-磷酸脱氢酶(glucose 6 phosphate dehydrogenase,G6PD)的催化下产生的供氢体烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate,NADPH)参与调节多种代谢反应和维持细胞氧化还原平衡;第二阶段是PPP 的非氧化途径,产生的5-磷酸核糖为核苷酸和氨基酸生物合成提供前体物质。因此,当PPP 增强时可增强细胞抗氧化应激能力,并为细胞提供快速增殖所需的底物,促进细胞增殖,抑制细胞凋亡。在人肺微血管内皮细胞和特发性肺动脉高压患者的 Fibs 中 PPP 显著增加[13,25],在多个 PH 模型中,肺血管细胞的PPP通路也上调[26-28]。
G6PD 是 PPP 途径的限速酶。在 PH 中,G6PD 上调或过度活化促进肺血管细胞(PASMCs、ECs 和Fibs)增殖且抑制细胞凋亡[9],导致肺血管重塑。其中,Chettimada 等[26]的研究表明,低氧时 G6PD 的活性和表达均增加,通过上调Sp1 和缺氧诱导因子1,促进细胞合成收缩力较小的蛋白(心肌素和平滑肌22α)以及增殖性更高的蛋白(细胞周期蛋白A 和磷酸化组蛋白H3),从而促进PASMCs 增殖。另外,G6PD 活性的增加还影响肺动脉收缩蛋白,引起肺动脉收缩[29]。使用G6PD 的竞争性抑制剂6-氨基烟酰胺,可下调离体牛肺动脉的收缩蛋白,并抑制肺血管炎症[30]。脱氢表雄酮具有较强G6PD抑制活性,可抑制缺氧和野百合碱诱导的PH 模型动物肺血管阻力升高、肺血管重构和心力衰竭[31-33]。一项3 期临床试验(NCT00581087)表明,脱氢表雄酮可改善慢性阻塞性肺病相关PH 患者的6 分钟步行距离和肺血流动力学异常[34]。这些研究均指出G6PD 可能作为PH的潜在治疗靶点。
G6PD 的产物NADPH 具有抗氧化性能,可用于谷胱甘肽/硫氧还蛋白的再生。值得注意的是,在PH中,过量的NADPH 可促进NADPH 氧化酶活化,产生过氧化氢和活性氧类,促进疾病进展[9]。此外,NADPH 是脂肪酸代谢和脂质合成所必需的物质,参与形成增殖细胞的膜,促进细胞增殖。NADPH 还调控触发凋亡的细胞周期酶caspase 的活性,在抑制细胞凋亡方面起到关键作用[9]。总的来说,抑制NADPH 氧化酶或抑制NADPH 的过度生成可能成为治疗PH的有效方法。
2 PH中的脂肪酸代谢重编程
2.1 脂肪酸合成 脂肪酸合成代谢可增加脂质大分子,为快速增殖的细胞提供所必须的能量和中间代谢物,用于新膜生产、细胞内转化和信号传导,驱动细胞出现增殖和抗凋亡表型。脂肪酸合成酶是脂肪酸合成的关键酶,在快速增殖的细胞中表达上调。在低氧的PAECs 和PASMCs 及PH 动物模型中脂肪酸合成酶的表达及活性升高,抑制其在PAECs 中的活性则可以减少血管内皮生长因子的表达,并增加内皮型一氧化氮合成酶的表达,改善PAECs 的功能[35];抑制其在 PASMCs 中的活性可促进凋亡并降低细胞增殖能力;在PH 动物模型中,抑制脂肪酸合成酶的活性则减轻肺血管重塑、降低右心室压力及延缓右心室肥厚[35-36]。这些研究认为抑制脂肪酸合成酶的表达/活性具有治疗PH的潜力。
2.2 脂肪酸氧化 脂肪酸代谢的另一个过程是脂肪酸β 氧化(fatty acid oxidation,FAO),该过程可生成ATP 和乙酰辅酶A。值得注意的是,FAO 和葡萄糖氧化存在一个相互调控机制,即Randle循环(又称葡萄糖-脂肪酸循环),指当葡萄糖氧化增加时,FAO就会被抑制,反之亦然。Randle 循环中糖脂代谢相互调控,以保障供给线粒体的葡萄糖和脂肪酸间的相对平衡[37]。
目前,研究者对FAO 在PH 中的作用有不同见解。一方面,有研究指出内皮功能及右心脂质代谢正常状态的维持需要足够的FAO,PH 中FAO 减少,抑制FAO 将进一步加重ECs 功能障碍和右心室脂质沉积,将促进PH 进展[38-41];另一方面,有研究表明抑制FAO改善肺血管重构和右心室功能,缓解PH的发生发展。Sutendra 等[42]的研究指出,抑制 FAO 可增加PASMCs 葡萄糖氧化效率,从而改善动物模型的肺血管重构。对于右心室来说,正常情况下,心肌细胞主要依赖 FAO 产生 ATP,约占 60%~90%[43]。PH患者和动物模型的右室心肌细胞FAO 减少,右室存在长链脂肪酸和甘油三酯蓄积,循环游离脂肪酸和长链酰基肉碱水平升高,所以目前认为心肌脂毒性可能是由 FAO 受损和脂质运输增加导致[40-41,44]。研究表明,右心室FAO 受损促进PH 的进展,而在动物模型中使用部分脂肪酸氧化抑制剂曲美他嗪或拉诺拉嗪,虽然进一步抑制FAO,但右室肥厚及PH 进展却可得到改善[8,45]。而且一期临床实验也证实拉诺拉嗪治疗动脉型肺动脉高压(pulmonary arterial hypertension,PAH)安全和耐受性良好,它可以改善患者的右心室结构和功能(NCT01174173)[46]。这可能是由于FAO产生与葡萄糖氧化等量的ATP需要多消耗12%的氧气[47],而在右心室肥厚缺血的情况下,无法维持FAO 对氧气的需求,导致FAO 下降。药物干预进一步抑制FAO 从而增加葡萄糖氧化,使右心室产能增加,或有利于右心室功能的改善。总之,目前的研究显示FAO 是有潜力的PAH 治疗靶点,但FAO具体如何调节PH 中肺血管重构和右心室功能仍有待进一步研究。
3 PH中的氨基酸代谢重编程
3.1 谷氨酰胺代谢 细胞内的谷氨酰胺通过谷氨酰胺酶脱氨形成谷氨酸,再经谷氨酸脱氢酶形成α-酮戊二酸进入三羧酸循环,并伴有NADH/NADPH 的生成,因此该代谢途径既可调节三羧酸循环,也可调控细胞内的氧化还原稳态。而且由于它为嘌呤、嘧啶和蛋白质的生物合成提供氮,其对于增殖的细胞必不可少[48]。多项研究显示,Yes 相关蛋白 1(Yesassociated protein 1,YAP1)/具有PDZ 结合域的转录共刺激因子(transcriptional co-activator with a PDZ binding domain,TAZ)轴激活后可上调谷氨酰胺酶,促进谷氨酰胺分解,导致了细胞的过度增殖和迁移,研究指出YAP1/TAZ 活化可能是肺血管重塑的必经过程[49-50]。通过药理方法靶向抑制YAP1 或谷氨酰胺酶,可观察到谷氨酰胺分解减少,肺血管重塑减轻,PH 得到改善[51]。此外,右心室肥厚也与谷氨酰胺分解相关,有研究观察到肥厚的右心室中c-Myc-Max 通路激活可导致谷氨酰胺分解增加,产生的能量可使细胞快速增长。而在体抑制谷氨酰胺分解可恢复PDH的活性,使葡萄糖氧化增强,从而改善右心室肥厚和心脏功能[52]。目前的研究显示,谷氨酰胺分解在PH 中增强,可促进肺血管重塑及右心室肥厚,靶向谷氨酰胺分解通路,尤其是靶向谷氨酰胺酶/YAP1或许有望用于PH的治疗。
3.2 精氨酸及甘氨酸代谢 精氨酸可在一氧化氮合成酶的催化作用下生成一氧化氮(nitric oxide,NO),而精氨酸酶可与一氧化氮合成酶竞争共同底物L-精氨酸,生成鸟氨酸及尿素,从而使NO 生成减少;同时,不对称二甲基精氨酸也抑制一氧化氮合成酶,从而抑制NO 的生成。精氨酸代谢与PH 高度相关,PAECs 中精氨酸代谢酶和不对称二甲基精氨酸水平增加,将导致NO 生成减少和血管内皮功能紊乱,从而促进疾病的发生发展[53-55]。添加外源性的L-精氨酸可抑制精氨酸酶活性,增加NO 生成,从而减轻细胞氧化应激和内皮功能障碍,因此研究者认为补充L-精氨酸可用于改善PAECs功能和 PH 进展[56]。此外,甘氨酸代谢也参与PH 的病理生理过程,与内皮功能障碍密切相关。研究证实,BolA 家庭成员3(BolA family member 3,BOLA3)对于维持PAECs 甘氨酸稳态非常重要[57],敲除BOLA3可下调 ECs 的甘氨酸裂解系统的表达,从而提高肺组织甘氨酸水平并促进PH的发生。同时,该研究表明过表达BOLA4可以防止PH 的发生,但可被外源性给予的甘氨酸所逆转。目前,精氨酸和甘氨酸代谢在PH 中的作用仍不十分清楚,机制研究还局限在ECs,虽然在动物模型中调节这些氨基酸的代谢表现出对PH 的治疗作用,但具体作用机制仍待进一步研究说明。
4 基于PH代谢重编程的诊断方法
随着PH 代谢重编程的研究不断进展,早期、无创的诊断技术成为可能。其中,基于Warburg效应中葡萄糖摄取增加的特点,出现了一种代谢成像诊断方法。相较于每摩尔葡萄糖完全氧化可生成32 分子ATP,Warburg效应中,每摩尔葡萄糖只能产生2分子ATP,为了维持能量平衡,葡萄糖摄取增加,而这可以通过正电子发射计算机断层扫描术(positron emission computed tomography,PET)测定特定部位[18F]-氟代脱氧葡萄糖(fluorodeoxyglucose,FDG)来检测[58-59]。在 PAH 患者和动物模型中,该诊断方法的有效性已得到证实[59-60]。研究表明PAH 患者肺部和右心室的FDG 的摄取量显著高于健康对照组,但肺部的FDG 的摄取量与肺血管血流动力学及6 min步行距离无显著相关性[61],而右心室FDG 的摄取量不仅可以反映负荷大小[60],还可间接反映右心室的功能状态[58]。该诊断方法使用方便且无创,为实时定量测量代谢重编程提供可能,可用于评估药物治疗效果和疾病预后,以补充目前的血流动力学检查。在临床,FDG-PET 已在其他疾病的诊断中投入使用[60],因此用于PH 的诊断指日可待。另外,由于心脏四维血流磁共振成像可以提供更先进的功能性血流动力学信息,若与FDG-PET结合甚至耦合,可能为PH患者提供更好的诊断和预后评估方法[62,63]。
循环代谢物检测亦有望成为PH 的诊断手段之一。代谢组学技术,如超高效液相色谱-质谱技术,只需少量样本,即可识别出差异代谢物。通过这些代谢物可以区分生理和病理状态,并预测临床结果。目前,PH 中的代谢组学研究已取得一定进展,美国
国家心脏、肺和血液研究所的肺血管疾病计划(Pulmonary Vascular Disease Phenomics,PVDOMICS)和英国PAH 国家队列研究,通过高通量筛查患者血浆并结合其他诊断工具,正在收集特定PH 类型和不同疾病阶段的信息[64-65]。通过对患者和正常人血浆代谢物进行检测分析,目前已发现多种差异表达的血浆代谢物,如某些microRNA、三羧酸循环代谢物、多胺、脂质[65]、酰基肉碱[44,66]、氨基酸[65]等。这些代谢物的表达水平与血流动力学改变相关[67],代谢物的显著改变还能预测死亡风险[65]。下一代代谢成像和代谢组学生物标志物将使PH 的早期诊断成为可能。然而,更好地理解PH 的病理机制,大规模分析更加精准的临床标志物,以及消除高成本,才能使这些诊断方法得以成功实施。
5 靶向PH代谢重编程的新药物
5.1 二氯乙酸盐 PDK 在PH 患者的肺血管和右心室中转录上调,是PDH 的抑制因子,当PDH 被抑制时,其底物丙酮酸无法进入三羧酸循环,因此PDK是葡萄糖氧化的关键抑制因子,其表达或活性增加可促进 Warburg 效应形成[17,68]。二氯乙酸盐是一种小分子丙酮酸类似物,通过结合PDK N 端保守的变构位点对其起到抑制作用。由于正常组织中PDK不活跃,二氯乙酸盐对异常组织相对特异,对正常心脏和血管细胞影响不大[69-70]。目前二氯乙酸盐已临床应用于儿童乳酸性酸中毒[71]和成人多形性胶质母细胞瘤[72],主要毒性为可逆性周围神经病变[73-74]。研究表明,二氯乙酸盐治疗通过增加线粒体内的葡萄糖氧化,可防止炎症细胞积累、减轻血管重塑、恢复正常QTc 间期及部分恢复右心室收缩性[9,45,75-76],改善了多种动物模型的 PH[68-70,77]。临床试验证实,二氯乙酸盐可降低平均肺动脉压力和改善右心功能,缓解PAH,适当的剂量在患者中耐受较好[78]。同时,该研究指出,二氯乙酸盐对部分PAH患者无效,可能与沉默调节蛋白3和解偶联蛋白2的功能性异构有关,这些异构体通过降低PDH的功能导致二氯乙酸盐无法实现治疗作用。总的来说,二氯乙酸盐是一种极有潜力的PH 治疗药物,若可识别沉默调节蛋白3 和解偶联蛋白2 功能性异构的患者,将实现更加精准的治疗。
5.2 拉诺拉嗪 拉诺拉嗪是一种哌嗪衍生物,能选择性抑制晚期钠电流,高浓度的拉诺拉嗪能抑制FAO[79]。由于 FAO 与葡萄糖氧化间存在 Randle 循环,拉诺拉嗪通过抑制FAO可以促进葡萄糖氧化,此外,它还能激活PDH 进一步使葡萄糖氧化增加。2006年拉诺拉嗪被美国食品和药物管理局批准用于治疗慢性心绞痛和心律失常[80-82]。在PH 的研究中,拉诺拉嗪被证实通过抑制右心室的电重塑和结构重塑缓解PH 中的第一种类型,即PAH[83-84]。一些小的单中心临床研究最近已经完成,一项研究报告了12例PAH 患者(6例拉诺拉嗪和6例安慰剂),短期给予患者拉诺拉嗪并随访12 周,显示该药物安全和耐受性良好,不良反应较小(NCT01757808)[85]。另一项研究共有11 例有症状的PAH 患者参与,研究显示,拉诺拉嗪连续治疗3 个月可以改善症状、右心室结构和功能,但不影响血流动力学(NCT01174173)[46]。目前PAH 中拉诺拉嗪的研究已进入2 期临床试验(NCT01839110 和NCT02829034),结果尚未发表[86]。右室功能是影响PH 预后的关键,而拉诺拉嗪可通过改善心肌细胞脂代谢缓解右室重构及功能障碍,因此,拉诺拉嗪对于PH 尤其是存在右室功能障碍的患者具有重要临床价值。
6 展望
尽管PH 中的代谢研究经历多年的发展有很大进展,但代谢重编程的作用机制仍不十分清楚。目前主要集中在代谢酶的研究,而代谢中间产物的研究相对较少。肺血管细胞增殖需要新合成核酸和细胞膜等,而核苷酸和胆固醇代谢途径中代谢酶的研究仍是空白。氨基酸代谢目前主要涉及谷氨酸、精氨酸和甘氨酸,其他氨基酸的研究非常少。此外,虽然机制研究取得一定成果,但实际应用到临床的药物有限,这也归因于PH 复杂的病理生理机制。PH 的代谢重编程涉及多条代谢途径,或许靶向多条代谢途径可显著提高PH 治疗效果。由于代谢途径间存在相互调节和交差点(图1),因此寻找调节多条途径的关键靶点可能为PH 治疗带来新的可能。除了PH 治疗外,如何实现PH 早期预防及诊断也值得深入探究;虽然很多代谢酶在PH 中高表达,但目前并没有实现临床转化使PH 患者获益,这也将是未来重要的研究任务。
猜你喜欢
World Journal of Pediatrics(2023年11期)2023-12-11 01:35:46
知识窗(2020年4期)2020-04-24 09:21:24
中国现代医药杂志(2020年12期)2020-02-06 06:32:16
心肺血管病杂志(2019年9期)2019-12-09 08:34:02
电子测试(2018年11期)2018-06-26 05:56:52
环球时报(2017-04-17)2017-04-17 07:04:56
海外星云(2016年23期)2017-01-06 17:48:54
——艾玛·莫拉诺的117岁生日
海外星云(2016年23期)2017-01-06 03:28:59
中外医疗(2015年11期)2016-01-04 03:58:50
医学研究杂志(2015年12期)2015-06-10 06:57:46
