
4-(3-氯苯乙基)呱啶的简便合成
2021-12-02 12:30:06张小玲
合成化学 2021年11期
奉 强, 常 波, 何 冰, 张小玲
(1.成都师范学院 a. 化学与生命科学学院 b. 功能分子结构优化与应用四川省高校重点实验室,四川 成都 611130)
呱啶为N-杂六元饱和化合物,衍生物种类繁多,是一类重要的有机化工中间体,在药物工业应用广泛。研究人员通常在呱啶环氮原子上或3-/4-位进行取代衍生,已成功开发众多呱啶药物,如局麻药物[1]、抗组胺药[2]、止痛药[3]、止泻药、治疗II型糖尿病[4-5]、抗逆转录病毒药物aplaviroc类似物及帕金森药等[6-7]。此外,芳基哌啶在抗菌药物方面还表现出良好的应用前景[8]。
近年来,研究人员开展了基于呱啶衍生物的新药,尤其是抗肿瘤药物的创制探索[9-10]。为改善药物分子的理化性质,通常会在芳基呱啶衍生物苯环上引入氯原子,如在苯基引入3-位氯原子(Scheme1,7)。

Scheme 1
合成该类化合物一般采用钯碳催化加氢还原方法。本文以廉价的呱啶化合物N-Boc呱啶-4-甲醇(1)和间氯苄氯(3)为原料;1经Swern 氧化得到N-Boc呱啶-4-甲醛(2);3与三苯基膦反应得到膦鎓盐(4);2与4经Wittig反应得到成烯产物(5);5经钯碳加氢、氯化氢-二氧六环溶液作用下脱Boc保护基反应得到目标化合物4-(3-氯苯乙基)呱啶盐酸盐(7, Scheme 1)。
1 实验部分
1.1 仪器与试剂
Bruker Mercury-400 MHz型核磁共振仪(DMSO-d6和CDCl3为溶剂,TMS为内标);Agilent 1200型高效液相质谱仪[色谱柱:Kinete C18柱(100 mm×4.6 mm×2.6 μm)流动相:A相为乙腈,B相为0.05%TFA/水溶液,流速1 mL/min;检测波长214 nm;柱温40 ℃]。
钯碳(干钯碳,含量为10%)、钯碳(含水量50%,钯含量5%)、二氯化钯,国药上海化学试剂公司;氢氧化钯、氧化铂、N-Boc呱啶-4-甲醇、三苯基膦、间氯苄氯,分析纯,上海毕得医药公司;草酰氯,分析纯,北京伊诺凯科技有限公司;其余试剂均为分析纯,成都科龙化工试剂公司。除二氯甲烷经氢化钙以及四氢呋喃经金属钠除水处理外,其余皆无须处理直接使用。
1.2 合成
(1)2的合成
在干燥的2 L四颈圆底烧瓶中放入强磁力搅拌子和500 mL干燥的二氯甲烷,瓶口装上干燥管、恒压滴液漏斗和-80 ℃低温温度计。一次性加入71.5 g(0.563 mol)新蒸草酰氯后,将混合体系冷至-60~-70 ℃,滴加二甲亚砜110 g(1.41 mol)的二氯甲烷(150 mL)溶液,滴毕(20 min),剧烈搅拌20 min ;滴加N-Boc呱啶-4-甲醇75 g(0.349 mol)的二氯甲烷(150 mL)溶液,滴毕(20 min),剧烈搅拌20 min;于-50 ℃滴加三乙胺190 g(1.88 mol)的二氯甲烷(150 mL)溶液,滴毕,保温20 min;撤去冷浴,自然升温至0 ℃。将混合体系倒入1 L冰水中,搅拌,分液,水相用二氯甲烷300 mL萃取,合并有机相,用纯水洗涤,真空干燥,减压蒸除溶剂得265.1 g,收率88%。
(2)4的合成
在2L单颈圆底烧瓶中加入138 g(0.6 mol)三苯基膦,64 g(0.4 mol)间氯苄氯和600 mLN,N-二甲基甲酰胺,搅拌使其溶解;氮气保护,回流反应3 h,其间体系析出大量白色不溶固体。趁热过滤,滤饼依次用50 mLN,N-二甲基甲酰胺和100 mL乙醚洗涤,于60 ℃真空干燥5 h得4146.0 g。产品不必纯化,直接用于下一步反应
(3)5的合成
将264.0 g(0.30 mol)和4135.0 g(0.34 mol)溶解干燥的900 mL四氢呋喃中,氮气保护下,将混合体系冷却至0 ℃,分批加入氢化钠14.8 g(60%分散在矿物油中,0.37 mol),加毕,撤去冷浴,自然升温至室温,搅拌反应过夜(TLC监测)。将反应物倒入500 mL冰中,搅拌至冰融化后加入500 mL冷石油醚,继续搅拌30 min;滤去不溶物,滤液分液得到有机相,滤饼用100 mL冷石油醚洗涤,水相用300 mL石油醚萃取,合并所有有机物,减压除去溶剂得淡黄色油状液体;剧烈搅拌下,将油状液体分散于800 mL冷石油醚中,搅拌30 min;过滤,滤饼用100 mL冷石油醚洗涤,合并滤液和洗液,真空除溶得淡黄色油状液体(5)74.3 g,收率77%。
(4)6的合成
将572.0 g(0.224 mol)和干钯碳7.0 g(含10%钯)加入600 mL 1,4-二氧六环中,分别用氮气和氢气置换3次,在氢气氛围中进行加氢反应4 h(TLC监测)。滤除钯碳,滤液减压蒸除溶剂得油状液体(6)69.6 g,收率96%;1H NMR(300 MHz, DMSO-d6)δ: 7.43~7.20(m, 4H), 3.72(d,J=12 Hz, 2H), 3.28(t,J=10.2 Hz, 2H), 2.60(t,J=6.6 Hz, 2H), 1.83(m, 2H), 1.49(m,4H), 1.42(m, 1H), 1.39(s, 9H);13C NMR(75 MHz, DMSO-d6)δ: 159.73, 130.88, 129.60, 128.50, 127.96, 125.26, 124.41,79.87, 47.68, 36.61, 35.83, 32.65, 29.28, 18.81; MS(EI)m/z: 324.2{[M+H]+}。
(5)7的合成
将667.2 g(0.208 mol)溶解在750 mL二氧六环中,冰盐浴冷却至0 ℃,缓慢滴加4 mol/L的氯化氢二氧六环溶液250 mL,滴毕,撤去冰浴,自然升温至室温,搅拌下反应16 h(TLC检测)。减压过滤,滤饼用300 mL无水乙醚洗涤后自然干燥10 min,于40 ℃真空干燥4 h得白色固体747.0 g,收率87%, LC-MS检测纯度为96.6%;1H NMR(300 MHz, DMSO-d6)δ: 9.03(s, 1H), 8.81(s, 1H), 7.33~7.17(m, 4H), 3.21(d,J=12.0 Hz, 2H), 2.78(t,J=10.2 Hz, 2H), 2.64(dd,J=24.0 Hz, 6.6 Hz, 4H), 1.83(t,J=5.8 Hz, 2H), 1.47(d,J=6.0 Hz, 2H), 1.29(m, 1H);13C NMR(75 MHz, DMSO-d6)δ: 154.73, 140.98, 139.62, 135.06, 129.41, 128.50, 127.96, 79.67, 36.61, 35.83, 32.65, 18.84; MS(EI)m/z: 224.1{[M+H]+}。
合成7时,文献方法一般以乙醇作溶剂,经钯碳催化加氢完成反应[11-13]。但实验中发现,反应顺利得到目标化合物5后,不能得到预期化合物6,只能得到6和6的脱氯混合物。为此,本文探究化合物5加氢合成化合物6的工艺条件。通过考察不同的催化剂、不同溶剂对反应产物的影响,最终筛选出最佳条件为:以二氧六环为溶剂,含量10%干钯碳催化加氢,室温下反应3 h。
5中碳碳双键的还原,最常用的方法就是在极性溶剂(常为甲醇或乙醇等质子性溶剂)中加氢反应。故选用实验室中常用钯碳催化剂(5%钯碳,含水量为50%),考察不同催化剂用量和不同质子性溶剂中催化加氢情况,结果得到加氢和脱氯的混合产物,结果见表1。由表1可知,反应一旦发生,就有加氢和脱氯产物生成,而不会只发生单一的加氢不脱氯反应。其原因可能是碳碳双键与苯环共轭,增加了碳碳双键的稳定性进而降低了其加氢活性,使得与芳环共轭碳碳双键加氢时苯环上的碳氯键也发生了加氢脱氯。

表1 催化剂用量和溶剂对6收率的影响
以乙醇作溶剂,考察不同催化剂对反应产物的影响,结果见表2。由表2可知,以乙醇作溶剂,不同催化剂对6的收率有较大影响。在6种钯催化剂中,反应均给出不可忽视含量的脱氯产物(脱氯产物与加氢脱氯产物之和)。其中相对较好的是含量10%的干钯碳,加氢目标物6的收率达到近50%(No.4)。可能是氢氧化钯活性太高,6的收率仅有26%,多数发生了脱氯反应(No.6)。虽然换用二氧化铂和铂碳催化氢化能达到较多加氢不脱氯产物的结果(No.7、 No.8),但其相对较高的价格对反应经济性有不利影响。

表2 催化剂类型对6收率的影响
由于其它钯催化剂催化加氢效果不如含量10%干钯碳,因此重点考察了10%干钯碳在多种有机溶剂中的催化效果,结果见表3。由表3可知,在含量10%干钯碳催化下,5在众多溶剂中反应,几乎都能发生加氢和脱氯的反应,尤其在乙酸乙酯中,脱氯的反应产物远多于加氢产物(No.6)。但以1,4-二氧六环作溶液,加氢反应相对容易发生,并且几乎没有检测到加氢脱氯反应产物的存在(<1%, No.12)。
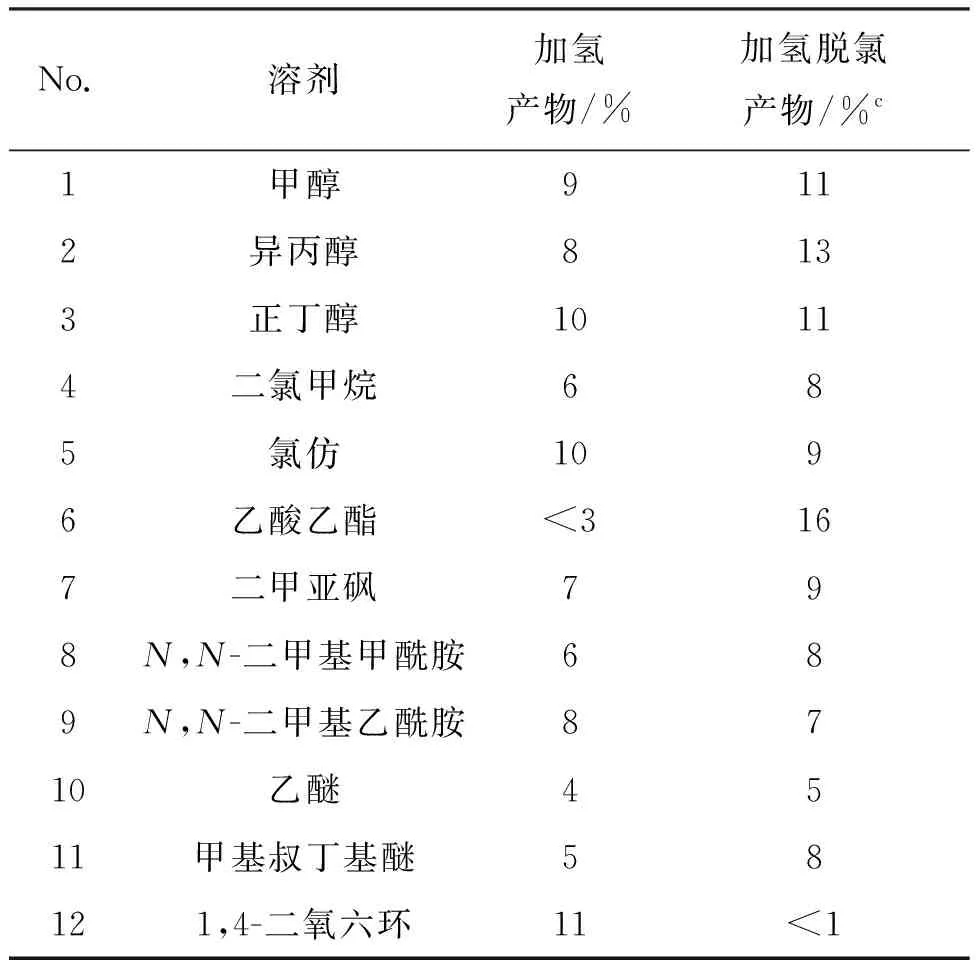
表3 不同溶剂对6收率的影响
为此,以1,4-二氧六环为溶剂,进一步考察含量10%干钯碳催化剂的用量及反应时间对6收率的影响,结果见表4。由表4可知,以1,4-二氧六环为溶剂,含量5%和10%干钯碳作催化剂,加氢反应都能顺利进行,得到加氢产物6为主产物的实验结果(No.4、 No.6、 No.7),随着反应时间的延长,脱氯产物逐渐增加(No.3、 No.4和No.7、 No.8)。

表4 催化剂用量和反应时间对6收率的影响
以N-Boc呱啶-4-甲醇和间氯苄氯为起始原料,经Swern 氧化、Wittig反应、钯碳催化加氢等反应合成了药物中间体4-(3-氯苯乙基)呱啶盐酸盐,反应总收率达到56%,产物纯度高达96.6%。通过对关键步骤加氢反应进行条件优化,筛选了一条合成7的廉价途径,为该中间体的放大生产提供了参考。
猜你喜欢
佳木斯大学学报(自然科学版)(2020年1期)2020-02-28 05:30:52
上海化工(2018年10期)2018-10-31 01:21:06
中成药(2017年12期)2018-01-19 02:06:26
化工管理(2017年32期)2017-11-24 06:20:21
绿色科技(2017年8期)2017-05-22 21:06:55
农产品加工(2017年6期)2017-05-09 18:04:52
兵工学报(2014年10期)2014-06-27 05:41:50
中成药(2014年9期)2014-02-28 22:28:43
化工生产与技术(2014年5期)2014-02-27 13:42:02
东南大学学报(自然科学版)(2012年3期)2012-06-28 03:59:00
