
析氢反应中氮掺杂石墨烯负载金属单/双原子催化活性起源
2021-11-19 09:23张诗诗秦棪阳苏亚琼
储能科学与技术 2021年6期
张诗诗,秦棪阳,苏亚琼,2
(1西安交通大学化学学院,陕西西安 710049;2埃因霍温理工大学化学工程与化学系,荷兰埃因霍温 5600MB)
氢气具有含量丰富、可再生等特点,可作为取代化石燃料的新型清洁能源。电催化析氢反应(HER)发生在水分解反应中,可实现高纯度、高产量的产氢效果,是一种可持续的安全可靠的氢气生产技术。然而HER 通常需要较大的过电位(η),为降低过电位,减少电能消耗,设计高效低廉的HER 电催化剂是当前制氢技术的发展趋势。近年来,基于过渡金属的电催化剂被广泛研究,过渡金属具有独特的d电子结构,在电催化反应中表现出优异的催化活性。
单原子催化剂(SAC)仅具有单个活性位点,可实现100%原子利用率,是电催化中极具潜力的贵金属催化剂替代品。通过实验以及理论研究,SAC 的催化活性已在多种电催化反应中得到证明[1-2],Xu 等人[3]利用理论计算证明了MN4/C 结构的SAC 在氧还原反应(ORR)中表现出高催化活性;Zhao 等人[4]表明SAC 在氮气还原反应(NRR)中也表现出高催化性能;HER 中,SAC 的高催化性能也得到了证明[5-7]。Cheng 等人[8]制备出Pt原子催化剂用于HER 中,实验结果表明相对于商业中使用的Pt/C 催化剂,SAC 具有高催化活性、高稳定性;Lei 等人[9]利用Ni 纳米粒子对Ni-N-C 结构的SAC 进行调控,使其在HER 中催化性能得到提升。由于SAC 的单活性位点,其催化性能具有局限性,具有双位点的双原子催化剂(DAC)被引入,DAC 中第二个活性位点对第一个活性位点的电子结构起到了调控作用,在NRR、ORR 等电化学反应中表现优异[10-11]。
为探明SAC、DAC 在HER 中的催化性能,本文主要选取了以三种过渡金属(Fe、Co、Ni)为活性中心的SAC、DAC,通过第一性原理计算对其结构稳定性、HER活性、电子结构做出了分析研究,为高效HER 电催化剂的研究提供理论支撑,并对新型HER催化剂的设计进行了展望。
1 计算方法
本文所有工作均使用基于密度泛函理论(DFT)的计算软件VASP。体系中波函数采用平面波基组的展开方式,赝势选择PAW,交换相关能使用广义梯度近似(GGA)的PBE表示。所有体系选取的平面波截断能为400 eV,k点抽样选择Monkhorst-Pack抽样法,k网格点为3×3×1。
为评估催化剂结构稳定性,引入结合能,其表达式为:Eb=Etot-Esupport-ETM,其中,Eb为体系结合能,Etot为整个结构的能量,Esupport、ETM分别为基底以及过渡金属原子的能量。结合能可表明各组成部分结合的紧密程度。

2 结果与分析
2.1 结构稳定性
本文选取Fe、Ni、Co 三种过渡金属元素作为活性中心,选取氮掺杂石墨烯(N-graphene)作为基底,设计了FeN3、FeN4、Fe2N6、CoN4、Co2N6、NiN4、Ni2N6七种原子催化剂。其结构为如图(1)所示的六方晶格体系,基矢为:a=b=14.76 Å,c=15 Å,真空层厚度为15 Å(1 Å=0.1 nm)。

图1 (a)FeN3、(b)FeN4、(c)Fe2N6结构的侧视及俯视图,橙色、蓝色、灰色小球分别为Fe、N、C原子Fig.1 The side and top view of the geometry optimization structures of(a)FeN3,(b)FeN4,(c)Fe2N6-graphene.Cray,blue,orange balls represent C,N,Fe atoms,respectively
为评估催化剂稳定性,我们将TM—N 键长及结合能置于表1 中。Chen 等人[10]曾提出2.00 Å 的TM—N 键长意味着SAC 结构良好的稳定性;键长越短表明原子结合越紧密,因此,从表1 可得本文所研究SAC、DAC 体系均具有良好的结构稳定性。此外,通过电荷密度差分,可以看出TM 和N 成键时,电荷从TM 转移到N 原子,进行了重新排布,聚集在TM—N 键上,意味着TM—N 为强化学键。

表1 各SAC、DAC结构中TM—N键长、结合能Table 1 The TM—N bond lengths and the binding energy of all the catalysts

图2 (a)CoN4、(b)Co2N6电荷密度差分图,C、N、Co原子分别用棕色、蓝色、红色小球表示,蓝色、黄色阴影分别代表电荷消耗和聚集Fig.2 The differential charge density maps of(a)CoN4,(b)Co2N6,Brown,blue,red balls represent C,N,Co atoms,respectively,blue and yellow shadows characterize the charge loss and accumulation
结合能进一步证明了本文催化体系的热稳定性,由结合能定义可知结合能值越负意味着结合能力越强。从表1 可看出DAC 相对于SAC 具有更高的结构稳定性,CoN4、FeN4稳定性较高,这在实验中已得到证明[13]。
2.2 HER催化活性
电催化析氢反应(HER)是在电极/电解质界面处发生的多步电化学反应过程,主要是通过还原质子(H+)生成H2:2(H++e-)→H2。其反应步骤分为两步,第一步反应为质子的吸附(Volmer反应),第二步为H2的脱附(Heyrovsky/Tafel反应)。
为保持较优的反应速率,HER 中氢原子的吸附和脱附需要达到良好的平衡[14]。H 原子吸附能是普遍采用的HER活性描述符[7,15],为研究在HER中所选取SAC、DAC 的活性,我们利用VASP 计算得出其吸附H的自由能变化(ΔG)。根据Sabatier原理,ΔG接近0时,HER催化效率达到最大,因此,高效HER 电催化剂对H 的吸附能力要保持适中水平。图3是HER在7种催化剂上进行时的自由能变化图,过电位已在图中标出。可以看出CoN4具有最小的过电位数值(-0.04 V);FeN3、FeN4、Fe2N6过电位分别为0.10、0.29 和0.36 V,HER 活性较好;Co2N6,Ni2N6,NiN4过电位较高(0.76、1.35、1.28 V),对H 吸附能力太弱,导致不理想的HER活性。Tour等人[16]在实验中合成了分散在N掺杂石墨烯上的单Co 原子催化剂(Co-NG),并证明在HER 中其过电位仅30 mV,这与本文理论计算所得结论相近。Lu 等人[6]通过改变N 配位数,对CoNx-gra(x=1~4)体系在HER 中催化性能进行了对比,从实验角度证明了CoN4-gra 具有最高的催化活性。Fe-Nx作为高效电催化剂活性中心,在NRR、ORR 等电化学反应中应用广泛,但少有报道将其应用于HER 中,本文DFT 计算结果表明FeN3、FeN4、Fe2N6三种以Fe 原子为活性中心的催化材料都具有作为HER电催化剂的潜能。另外,本文将SAC 与DAC 进行对比,利用DFT 计算证明: 对于HER, SAC 催化活性高于DAC。Nørskov 等人[15]发现金属表面引入H2O 分子层会使H 吸附能降低0.02 eV,因此H2O 分子质子化的微弱影响可以忽略。

图3 HER中各催化剂上能量变化图Fig.3 The free energy diagrams of HER on all catalysts studied.The overpotentials(η)are presented
为进一步研究活性的来源,我们对活性较好的CoN4,FeN3,FeN4,Fe2N6进行了电荷分析。CoN4态密度如图4(a)所示,通过态密度分析可得出各金属原子催化剂中金属的d带中心值:-2.24、-2.53、-2.73、-3.24 eV,由“d带中心理论”(d带中心值越高,意味着吸附强度越高)可得,CoN4具有最强的吸附能力,该结论与计算所得吻合。此外,如图4(b)所示,我们发现d 带中心值和HER 中ΔG(*H)具有近线性关系,因此,可以证明催化剂活性和金属活性中心有关,且d带中心值可作为HER中催化剂活性的描述符。

图4 (a)CoN4中Co的d轨道态密度图,(b)d带中心值和HER过电位的近线性关系示意图Fig.4 (a)Projected density of state(PDOS)projected on 3d orbitals of Co atom of CoN4.(b)The calculated linear relation of the ORR overpotential(-ηORR)as a function of d-band center(εd)values of Co atom
如图5 所示,为明确CoN4活性,我们对其吸附H 后结构的电荷分析做出了进一步讨论,由电荷密度差分可以看出H 吸附后,活性中心Co 原子将电子转移给H。图5(b)中可以看出H 被吸附后,Co 的d 轨道态密度图有新的峰值出现,新峰值分别代表成键轨道(σ)和反键轨道(σ*),这意味着Co 原子和被吸附H 之间具有相互作用,Co原子作为活性中心,其d 轨道与H 直接进行相互作用。
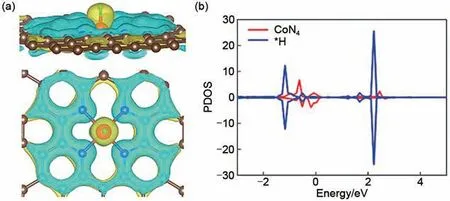
图5 CoN4吸附H后(a)电荷密度差分图,棕色、蓝色、红色、绿色小球分别代表C、N、Co、H原子,黄色和蓝色阴影分别代表电荷聚集和电荷消耗;(b)Co的3d轨道以及被吸附*H的1s轨道的态密度图Fig.5 (a)The differential charge density maps where the brown,blue,red,green balls denote the C,N,Co,H atoms,respectively;(b)The projected density of state(PDOS)projected on 3d orbitals of Co atom and the 1s orbital of adsorbed H of CoN4-H
3 结论
本文选取3种不同金属中心的SAC、DAC,通过DFT 计算分析讨论了HER 中各催化剂的活性,并对催化剂电荷结构进行了分析。结果表明CoN4吸附H步骤具有最接近于0的自由能变化,表现出最优异的催化性能,FeN3、FeN4次之(∆G=0.10,0.29 eV);DAC 在HER 中具有较差的催化活性;Ni 原子作为活性中心无法很好地催化HER 过程。通过电荷分析发现d带中心和催化活性之间存在着近线性关系;吸附H时金属活性中心和H原子之间具有较强的相互作用。
猜你喜欢
大学物理(2022年9期)2022-09-28
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
物理通报(2020年7期)2020-07-01
原子与分子物理学报(2015年3期)2015-11-24
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
河北科技大学学报(2015年5期)2015-03-11
无机化学学报(2014年4期)2014-02-28
无机化学学报(2014年3期)2014-02-28
