
2-溴-1,1,1-三氟-2-甲基丙烷的合成工艺研究
2021-11-18 05:35:26赵爱明倪秋洋
有机氟工业 2021年2期
赵爱明 倪秋洋
(1. 海门瑞一医药科技有限公司 研发部,江苏 海门 226121;2. 开美化学科技(南通)有限公司 研发部,江苏 南通 226100)
0 前言
三氟甲基(CF3)具有强吸电子性、亲脂性和稳定的C—F键(键能460 kJ/mol)等独特性质,将其引入到有机化合物中能够显著改变化合物的酸性、极性、可吸收性以及代谢稳定性[1-3]。含CF3的有机化合物正处于药物化学[4-6]、农药化学[7]以及材料科学等领域的研究前沿。2019—2020年,FDA批准的含氟小分子药物中有多个含有CF3,例如:全新靶点多发性骨髓瘤新药Selinexor(塞利尼索)、抗乳腺癌药Alpelisib(阿培利司)、预防遗传性血管性水肿发作的口服非甾体药物Berotralstat(贝罗司他),结构式见图1。另外,对生物活性分子三氟甲基化已成为改造其生物活性的一种有效手段, 在新型药物设计和农药开发中日益受到研究人员的重视[5]。如何向有机分子中有效引入CF3,具有非常重要的意义。目前构筑三氟甲基化合物有两种策略:1)直接三氟甲基化反应,底物直接与四氟化硫或三氟甲基化试剂(如Umemoto试剂和Togni试剂等)反应[5,8-9],引入CF3。该法最直接,但存在反应条件较苛刻、部分三氟甲基化试剂价格昂贵、毒性腐蚀性强、部分试剂未商业化或使用过程中污染大,且能直接反应的底物有限等缺点;2)三氟甲基砌块的反应[10-11]。此为间接法,将三氟甲基合成子经多步反应合成许多具有潜在生理、药理活性的复杂三氟甲基化合物[12-14]。

图1 FDA批准的部分含氟小分子药物
2-溴-1,1,1-三氟-2-甲基丙烷(1)是一种重要的含CF3砌块,可用于多种潜在药物活性化合物的合成,应用前景广泛[15-16]。文献报道[17],以2-溴-2-甲基丙酸为原料与SF4发生氟代反应制备1。该路线对生产设备要求高,氟化剂SF4价格昂贵,用量大,工业化的成本较高,且对环境污染大,不利于工业化生产。针对现有不足,本研究首次以1,1,1-三氟-2-甲基-2-丙醇(2)为原料,先与磺酰氯(3)反应生成磺酰酯(4),再置换溴化得到1,其结构经1H NMR及19F NMR表征。合成反应式如下:

通过试验发现:选择甲基磺酰氯(3a)与2反应生成甲基磺酰酯(4a),再溴化得1,收率较高。对4a及1的合成工艺进行了优化,确定了最佳反应条件。本法中间体4a无需提纯,直接进行下一步反应,操作方便,安全易行,设备要求不高,对环境友好,是一种高效合成2-溴-1,1,1-三氟-2-甲基丙烷的新方法。
1 试验部分
1.1 试验原料与仪器
1,1,1-三氟-2-甲基-2-丙醇,自制;4-二甲氨基吡啶,分析纯,上海阿拉丁生化科技股份有限公司;2.2 mol/L正丁基锂的正己烷溶液,试剂级,上海阿拉丁生化科技股份有限公司;四丁基溴化铵,分析纯,萨恩化学技术(上海)有限公司;溴化锂,化学纯,国药集团化学试剂有限公司。其余所用试剂均为工业级。
GC-2010气相色谱仪(OV-1色谱柱,30 m×0.25 mm×0.25 μm),日本岛津公司;AVANCE 300 MHz核磁共振仪(CDCl3为溶剂,TMS为内标),瑞士布鲁克公司;强力恒速搅拌机,1506B,常州新析仪器有限公司;Metrohm 848型电位滴定仪,瑞士万通;数显恒温油浴锅,W505,上海禾汽玻璃仪器有限公司。
1.2 试验过程
1.2.1 甲基磺酰酯(4a)的合成
四口烧瓶中依次加入1,1,1-三氟-2-甲基-2-丙醇(2)25.6 g(0.2 mol)、二氯甲烷200 mL、三乙胺30.4 g(0.3 mol)和4-二甲氨基吡啶1.2 g(0.01 mol),搅拌。N2保护下,冷却至0 ℃,滴加甲基磺酰氯(3a) 24.1 g(0.21 mol)与二氯甲烷25 mL的混合液。滴毕,30 ℃搅拌反应12 h,冷却,滴加水终止反应。静置、分层,有机层用饱和氯化钠水溶液洗涤,减压旋蒸得浓缩液,称重41.1 g,收率99.7%。1H NMR δ: 1.75(6H,s,OC(CF3)(CH3)2) ,2.99(3H,s,SO3CH3)。数据与文献基本一致[18]。
1.2.2 对甲基苯磺酰酯(4b)的合成
四口烧瓶中依次加入1,1,1-三氟-2-甲基-2-丙醇(2) 25.6 g(0.2 mol)和四氢呋喃100 mL,搅拌。N2保护下,冷却至0 ℃,滴加2.2 mol/L正丁基锂的正己烷溶液100 mL(0.22 mol)。滴毕,20 ℃反应30 min。N2保护下,冷却至-45 ℃,滴加对甲基苯磺酰氯(3b) 40.0 g(0.21 mol)与四氢呋喃40 mL的混合液。滴毕,缓慢升温至常温搅拌反应过夜。冷却,滴加氯化铵水淬灭反应。加入二氯甲烷,静置、分层,有机层用饱和氯化钠水溶液洗涤,减压旋蒸得浓缩液,称重46.7 g,收率82.7%。1H NMR δ: 1.76(6H,s,C(CH3)2),2.51(3H,s,PhCH3),7.42~7.91(4H,m,SO3Ph)。
1.2.3 2-溴-1,1,1-三氟-2-甲基丙烷(1)的合成
四口烧瓶中依次加入甲基磺酰酯(4a)61.9 g(0.3 mol)、丙酮200 mL、溴化锂39.1 g(0.45 mol)和四丁基溴化铵9.7 g(0.03 mol),搅拌。油浴加热升温至58 ℃回流反应14 h。冷却,加入水和二氯甲烷。静置、分层,有机层用饱和氯化钠水溶液洗涤,常压蒸馏收集67 ℃淡黄色透明液体,称重51.4 g,收率89.6%,纯度99.6% (GC面积归一法);1H NMR δ: 1.79(s,6H);19F NMR δ: -78.38(s,3F)。数据与文献基本一致[17]。溴含量为41.6%(理论值41.86%)。
84.7 g(0.3 mol)4b 代替4a,得黄色透明液体43.7 g,收率76.2%,纯度99. 2% (GC)。
2 结果与讨论
2.1 合成工艺的选择
羟基直接溴化,溴化剂一般选择三溴化磷或氢溴酸。因1,1,1-三氟-2-甲基-2-丙醇(2)的特殊结构,酸性条件下溴化更易发生脱水消除反应成烯或重排的副反应[20]。采用三溴化磷或氢溴酸直接溴化2,未能成功。因此,以2为原料,先与磺酰氯(3)反应生成磺酰酯(4),再置换溴化得2-溴-1,1,1-三氟-2-甲基丙烷(1)。其中3选择了3a和3b两种。通过试验发现,在敷酸剂三乙胺和催化剂4-二甲氨基吡啶(DMAP)存在下,3a与2发生反应生成4a,且收率高。而3b与2在三乙胺/DMAP体系下基本不反应。可能是3a与3b空间位阻不同所致。2可通过正丁基锂拔氢,再在低温下与3b反应生成4b。另外,4a溴化收率也高于4b溴化收率,这可能是因为4b溴化易发生消除副反应生成烯[19]。因此,为了避免使用易燃危险品正丁基锂,同时降低成本,选择收率相对高的路线——3a与2反应生成4a,再溴化得到1。
2.2 合成甲基磺酰酯(4a)的工艺优化
2.2.1 碱的选择
该反应为羟基的酰基化反应,常用吡啶、三乙胺等碱作为缚酸剂。不同碱体系对甲基磺酰酯(4a)收率的影响见表1。

表1 碱对4a收率的影响
由表1可见,使用吡啶既作缚酸剂又作溶剂,4a收率较低。先用正丁基锂拔氢,再在低温下与甲基磺酰氯(3a)反应,4a收率达90%以上。但正丁基锂易燃、高度危险,对设备安全要求高。单独使用三乙胺作缚酸剂,4a收率尚可。而添加催化剂DMAP能明显提高4a收率。这是因为DMAP结构上二甲氨基的孤电子对与吡啶母环共轭,激活了环上氮原子,提高了母环上氮原子的亲核取代活性,表现出显著的酰化催化效果[21],是一种新型高效酰化催化剂。因此,选择三乙胺/DMAP体系。
2.2.2 其他反应条件对收率的影响
其他反应条件对甲基磺酰酯(4a)收率的影响见表2。

表2 其他反应条件对4a收率的影响
由表2可见,采用滴加甲基磺酰氯(3a)的投料方式比采用滴加三乙胺的投料方式,4a收率高。这是因为采用滴加3a的投料方式,反应体系始终保持碱性,避免了消除及重排副反应的发生。从反应式看,它们的计量关系为n(2):n(3a)为1:1。由表2还可见,3a稍过量n(2):n(3a)为1.00:1.05时,有利于反应的进行,4a收率相对较高;继续增加3a用量,收率变化不大,反而造成3a的浪费,增加了原料成本。另由表2可见,反应温度≦30 ℃时,随着反应温度的升高,4a收率增加,且反应时间缩短,提高了生产效率。随着反应温度继续升高,4a收率反而下降。这是因为温度过低,反应不充分;温度升至30 ℃基本达到化学平衡,收率最高;温度过高,副反应增加,收率反而下降。故选择滴加3a的投料方式,n(2):n(3a)为1.00:1.05,30 ℃反应12 h。
2.3 合成2-溴-1,1,1-三氟-2-甲基丙烷(1)的工艺优化
该反应为置换溴化反应,考察了溴化体系、物料配比、反应温度和反应时间对反应的影响。
2.3.1 溴化体系的选择
不同的溴源及溶剂组成溴化体系,对1收率的影响见表3。
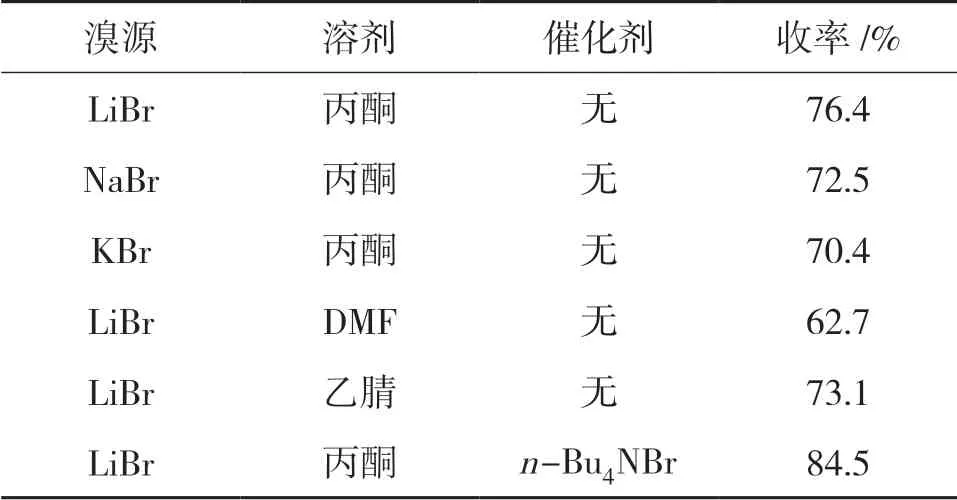
表3 溴化体系对1收率的影响
由表3可见,溴源选择LiBr相比于NaBr和KBr,收率更高;丙酮作溶剂相比于乙腈或DMF作溶剂,收率更高。这是因为LiBr相比NaBr和KBr在有机溶剂中溶解度更好,更有利于置换反应的进行。选择乙腈或DMF作溶剂时,置换反应的离去基团成金属盐未析出体系,绝大部分仍溶解在体系中,抑制了亲核取代反应的进行。另由表3可见,加入催化剂n-Bu4NBr,收率显著提高。这是因为n-Bu4NBr一方面催化了反应,另外又为反应提供了额外的溴源。因此,选择LiBr/n-Bu4NBr/丙酮作为置换溴化体系。
2.3.2 物料配比对收率的影响
物料配比对2-溴-1,1,1-三氟-2-甲基丙烷(1)收率的影响见表4。

表4 物料配比对1收率的影响
由表4可见,n(LiBr):n(4a)比值越大,收率也越高。当n(LiBr):n(4a)为1.5:1.0时,物质的量比继续增大,收率则无明显变化,而此时LiBr用量过大,造成浪费。另由表4可见,n-Bu4NBr作为催化剂和溴源,用量越多,收率越高。当n(n-Bu4NBr):n(4a)为1:10时,继续增加n-Bu4NBr用量,收率增加有限。从经济角度综合考虑,选择n(LiBr):n(4a)为1.5:1,n(n-Bu4NBr):n(4a)为1:10较适宜。
2.3.3 反应温度以及反应时间对收率的影响
反应温度以及反应时间对2-溴-1,1,1-三氟-2-甲基丙烷(1)收率的影响见表5。

表5 反应温度以及反应时间对1收率的影响
由表5可见,反应温度越高,置换反应越容易进行,收率越大,反应温度达到58 ℃回流时,收率最大,且反应时间最短,生产效率最高。温度过低,即使延长反应时间,反应仍不充分,收率较低。这是因为在回流的条件下,分子运动最剧烈,离去基团更易离去而形成碳正离子,另外LiBr溶解度变大,增加了体系内的溴源,促进了亲核取代反应的进行。另由表5可见,反应时间为14 h时,收率最大,此时基本达到化学平衡,再延长反应时间,收率基本不变,但影响效率,增加能耗。因此,选择58 ℃回流反应14 h为宜。
3 结论
首次以1,1,1-三氟-2-甲基-2-丙醇(2)为原料,先与磺酰氯(3)反应生成磺酰酯(4),再置换溴化得2-溴-1,1,1-三氟-2-甲基丙烷(1),其结构经1HNMR及19FNMR表征。通过试验发现选择甲基磺酰氯(3a)与2反应生成甲基磺酰酯:(4a),再溴化得1,收率较高。通过对4a合成工艺优化确定了最佳反应条件为:选择三乙胺/DMAP体系,滴加3a的投料方式,n(2):n(3a)为1.00 :1.05,30 ℃反应12 h,收率99.7%。无需提纯,直接进行下一步反应。通过对1合成工艺优化,确定了最佳反应条件为:LiBr/n-Bu4NBr/丙酮为置换溴化体系,n(LiBr):n(4a)为1.5 : 1.0,n(n-Bu4NBr):n(4a)为1:10,58 ℃回流反应14 h,收率89.6%。本法操作方便、质量可控,避免了使用文献中对环境污染大的四氟化硫、高度易燃危险品正丁基锂等,安全易行,对设备要求不高,且对环境友好,是一种高效合成2-溴-1,1,1-三氟-2-甲基丙烷的新方法,适合规模化生产。
猜你喜欢
应用化工(2023年1期)2023-02-16 10:56:46
橡胶科技(2022年9期)2022-12-12 05:26:53
河南化工(2020年11期)2020-12-10 05:43:20
分析化学(2019年3期)2019-03-30 10:59:24
浙江化工(2018年3期)2018-04-19 08:42:56
潍坊学院学报(2017年2期)2017-04-20 08:44:22
生物化工(2016年4期)2016-04-08 10:26:27
微生物学杂志(2015年1期)2015-12-26 08:18:01
橡胶工业(2015年9期)2015-08-29 06:40:46
山东工业技术(2015年6期)2015-07-27 00:53:22
