
PCR⁃RFLP法检测MTRR基因A66G多态位点的改进
2021-11-18 08:14何震宇顾取良陈瑜丽
分子诊断与治疗杂志 2021年10期
何震宇 顾取良 陈瑜丽
甲硫氨酸合成酶还原酶又叫5⁃甲基四氢叶酸⁃同型半胱氨酸甲基转移酶还原酶(5⁃methyltetrahy⁃drofolate homocysteine methyltransferase reductase,MTRR),在同型半胱氨酸重新甲基化为甲硫氨酸的过程中发挥着重要作用,是叶酸代谢网络的关键酶之一[1]。MTRR基因A66G 多态性(rs1801394 位点)会引起其编码的酶蛋白第22 位氨基酸由异亮氨酸变为甲硫氨酸(I22M),导致MTRR 的活性及同型半胱氨酸的甲基化速率降低,从而引起多种相关疾病的发生,如唇腭裂[2]、先天性心脏病、无脑儿或者脊柱裂[3]、自然流产[4]、早产[5]、H 型高血压[6]等。因此,检测MTRR基因A66G 多态性,对相关疾病的发病风险预测及临床研究均有重要意义。
目前有多种方法可检测MTRR基因A66G 多态性,包括扩增阻滞突变系统聚合酶链反应[7]、高分辨率熔解曲线[6]、SNaPshot 测序[8]及聚合酶链反应⁃限制性片段长度多态性(polymerase chain reac⁃tion⁃restriction fragment length polymorphism,PCR⁃RFLP)[2,9]等。本研究旨在对现有检测MTRR基因A66G 多态位点的PCR⁃RFLP 法进行改进,通过改变引物的设计,使得野生型等位基因A、突变型等位基因G 来源的PCR 产物均在多态位点外包含一个NdeI 识别位点,以期将它作为NdeI 酶切的内对照从而避免传统方法因NdeI 酶切不完全即残留PCR 产物时可能造成的基因型误判。
1 材料和方法
1.1 主要试剂
口腔拭子基因组提取试剂盒购自天根生化科技有限公司;2×EasyTaq PCR SuperMix(+dye)购自北京全式金生物技术有限公司;引物自行设计(序列见图1),委托生工生物工程股份有限公司合成;FastdigestNdeI 购自赛默飞世尔科技(中国)有限公司;DNA 分子量标准DNA MarkerⅠ购自北京庄盟国际生物基因科技有限公司。
1.2 研究对象
口腔拭子标本100 例,采自广东药科大学女性志愿者,平均年龄为(21.87±0.58)岁,事先均签署了知情同意书。
1.3 基因组DNA 的制备
准备医用消毒棉签,志愿者先用清水漱口,然后手持棉签伸进口腔,在腮帮子上反复擦拭10次,取出棉签,按试剂盒的说明书步骤提取基因组DNA。
1.4 引物设计
根据NCBI中MTRR基因(登录号:NG_008856.1)及dbSNP 数据库的相关序列信息,采用primer pre⁃mier 5.0 软件辅以人工修改设计引物。引物与模板的关系见图1,图中第1 位碱基为NG_008856.1序列中第6712 位碱基,图中第46 位碱基R(A/G)为多态位点碱基,上游引物Fo、下游引物Re 分别与基因组序列同向、反向。

图1 PCR 的模板序列、引物序列及引物与模板结合位置示意图Figure 1 Template sequence,primer sequence and primer⁃template binding location of PCR
1.5 PCR 扩增及产物的电泳鉴定
PCR 体系总体积50 μL,其中2×EasyTaq PCR Super Mix 25 μL、上下游引物各10 pmol、基因组DNA 1 μL,ddH2O 补足体积。PCR 反应条件:94℃预变性4 min;94℃30 s,60℃30 s,72℃30 s,35 个循环;72℃延伸7 min。PCR 产物使用1.5%的琼脂糖凝胶电泳进行鉴定。
1.6 PCR 产物的酶切分型
酶切反应体系30 μL,其中10×FastDigest Green Buffer 2 μL、FastDigestNdeI 1 μL、PCR 产物5~10 μL、ddH2O 补足体积,37℃水浴消化1 h,然后65℃水浴保温5 min 以灭活内切酶。酶切产物经3.0%的琼脂糖凝胶电泳进行检测,根椐限制性酶切图谱中的特征条带确定每份样品MTRR基因A66G 多态位点的基因型。
1.7 测序验证
挑选本法检出的3 种基因型样品,PCR 扩增其478 bp 的靶序列委托生工生物工程股份有限公司进行序列测定。
1.8 统计学处理
采用SPSS 24.0 统计学软件,计数资料以例数和频数表示,采用χ2检验进行Hardy⁃weinberg 遗传平衡吻合度检测以确定样本是否具有群体代表性,以P<0.05 为差异有统计学意义。
2 结果
2.1 MTRR 基因A66G 多态位点PCR 产物的电泳鉴定
PCR 产物理论大小为478 bp,结果符合预期。见图2。
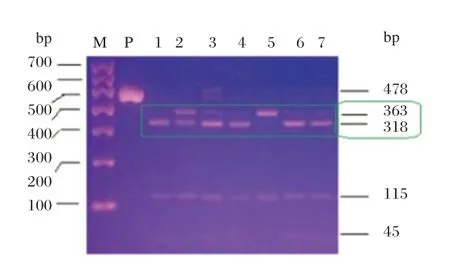
图2 改进的PCR⁃RFLP 鉴定MTRR 基因A66G 位点基因型之电泳图谱Figure 2 The electrophoresis map for identificating genotypes of A66G locus of MTRR gene by improved PCR⁃RFLP
2.2 MTRR 基因A66G 多态位点PCR 产物酶切图谱及基因型判断
电泳图谱里只出现363 bp 特征性酶切条带的为突变型纯合子GG 样品,如图2中的5 号样品;只出现318 bp 特征性酶切条带的为野生纯合子AA 样品,如图2中的1、4 号样品;363、318 bp 两种条带同时出现时,需区分杂合子AG 样品和因酶切不完全残留363 bp 条带的野生型纯合子AA 样品,图2中的3、6、7 号样品的363 bp 条带为弱带或痕量带,且还伴有痕量的478 bp PCR 产物残留带,应属于酶切不完全的情形,故其基因型均为野生纯合子AA,相反,图2中的2 号样品的两条条带均很明显,363 bp 条带亮度强于318bp 条带,样品的基因型为杂合子AG。见图3。
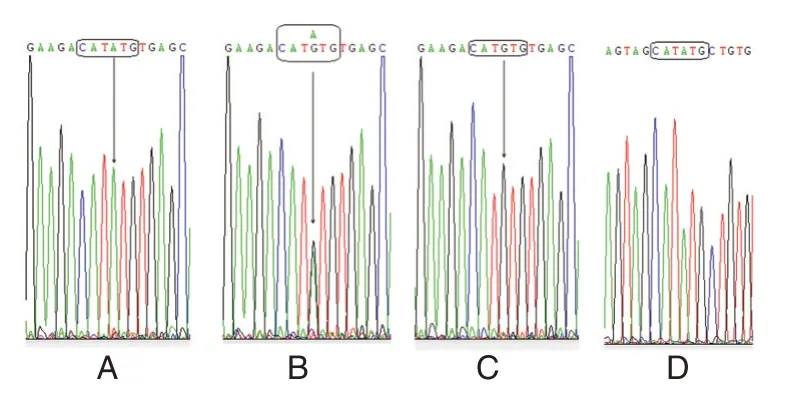
图3 MTRR 基因A66G 位点3 种基因型样品测序峰图Figure 3 Sequencing maps of three genotypes of A66G Locus of MTRR gene
2.3 Hardy⁃Weinberg 遗传平衡检验
本研究检测到AA、AG、GG 3 种基因型样品的例数分别为:57、34、9,相应的基因型频率分别为0.57、0.34、0.09。等位基因A 的频率为0.74,等位基因G 的频率为0.26。经Hardy⁃Weinberg 遗传平衡检验,χ2=1.355(P=0.508)。
3 讨论
采用PCR⁃RFLP 法检测MTRR基因A66G 多态位点的既有做法是通过错配上游引物及完全与模板配对的下游引物进行PCR,再用限制性核酸内切酶NdeI 消化PCR 产物制作酶切图谱分辨基因型。上游引物3′端倒数第3 位的错配碱基C 把基因组原序列AATRTG(R 为多态位点碱基)改变成PCR 产物中的序列CATRTG,当R 为野生型等位基因A 时,其可被NdeI 切割,反之,当R 为突变型等位基因G 时,其不能被NdeI 切割。该方法最早可追溯到Wilson A 等的研究[10],其所用引物扩增的PCR 产物长度为66 bp,使用NdeI 消化PCR产物,A 等位基因的PCR 产物会被切割成44 bp 和22 bp 的片段,G 等位基因的PCR 产物不会被切割,仍然保持66 bp 的条带,Rai V 课题组也曾使用这对引物进行研究[11]。由于上游引物需在多态位点附近引入错配碱基创造NdeI 酶切位点,其位置基本固定,一些研究人员通过改变下游引物的位置,获得了比66 bp 更长的PCR 产物用于分型,这些PCR 产物的 长 度为145 bp、151 bp、182 bp 不等[12⁃14]。所有这些方法获得的PCR 产物中除了多态位点相关序列CATRTG 外,没有额外的NdeI 识别序列,当野生型等位基因A 的PCR 产物没有被NdeI 完全消化的时候,其残留的PCR 产物很可能会造成样品携带G 等位基因的假象,基因型AA 样品会被当作基因型AG 样品对待,这种类似的弊端在针对其他基因多态位点的PCR⁃RFLP 分型法中已有报道[15]。
快速限制性核酸内切酶(简称快酶)FastDigestNdeI 消化PCR 产物的时间≥60 min,而绝大多数快酶可以在5~15 min 内完成对PCR 产物的消化,意味着NdeI 切割PCR 产物的效率相对较低,出现酶切不完全的概率较大,为了避免既有方法因NdeI酶切不完全可能导致的基因型误判,建立一种监测酶切程度的体系很有必要,在PCR 产物中引入内对照酶切位点能很好地监测PCR 产物是否酶切完全,在其它基因多态位点的检测中已有一定的应用[16]。本研究结果表明该设计既能很好地监测酶切是否完全,又能很好地通过特征性酶切条带来判断基因型。本研究对上游引物的长度也作出了相应的调整,既有研究的上游引物长度为20~26 nt 不等的普通引物,本研究的上游引物为长达45 nt 的长链引物,其中5′端的CCGGGAGCTGCATGTGT⁃CAGAGG 为附加序列,它来自通用测序引物pGEX3′,与模板毫不相关,只起到增加PCR 产物长度的作用,使得表征G 等位基因的特征性酶切条带(363 bp)与表征A 等位基因的特征性酶切条带(318 bp)在琼脂糖凝胶电泳图谱上有良好的区分度。下游引物的5′端也附加了一段与模板无关的序列,它来自通用测序引物CMV⁃F,旨在缩小上、下游引物退火温度的差异,从而保证PCR 的效率。
总之,本研究经过独特的引物设计,成功地在PCR 产物中引入了内对照酶切位点,且PCR 产物、表征A 等位基因的特征性酶切条带、表征G 等位基因的特征性酶切条带彼此之间具有良好的区分度,不仅可以监测酶切是否完全,而且在酶切不完全的情况下,能够根据两种等位基因的特征性酶切条带的具体情况判断样品的基因型,在相关的科学研究和临床检测中有一定的推广应用价值。
猜你喜欢
计算机应用与软件(2022年6期)2022-07-12
自然灾害学报(2022年2期)2022-05-10
智慧健康(2021年17期)2021-07-30
中国学校体育(2021年10期)2021-04-26
新课程·下旬(2018年9期)2018-11-14
电脑知识与技术(2017年10期)2017-06-05
电子技术与软件工程(2017年8期)2017-05-10
科技视界(2016年27期)2017-03-14
青少年科技博览(中学版)(2015年10期)2015-01-11
中学生物学(2008年6期)2008-08-29
