
α-丙氨酸Co3+配合物旋光异构及水溶剂效应的理论研究
2021-11-17 11:53:58彭国强徐锐英雷泽平杨清荟崔金玉赵丽红郝成欣王佐成
复旦学报(自然科学版) 2021年5期
彭国强,刘 军,徐锐英,雷泽平,杨清荟,崔金玉,赵丽红,郝成欣,王佐成,佟 华
(1. 白城医学高等专科学校 基础医学部,吉林 白城 137000; 2. 白城师范学院 物理学院,吉林 白城 137000; 3. 营口理工学院 基础教研部,辽宁 营口 115000)
氨基酸是构成蛋白质、酶等生物大分子的基本单元,其金属配合物是蛋白质、酶等生物大分子的活性中心,参与生命体的代谢及生化过程.氨基酸金属配合物的研究是生命科学和医学等领域的科研热点,具有重要的学术价值与应用价值[1].基于氨基酸金属配合物的重要作用,学者对其结构特性进行了大量的研究.文献[2-6]研究了气相和液相下Na+,K+,Ca2+,Cu2+等金属离子与几种氨基酸配合物的分子内质子迁移机理,探讨了不同构型的氨基酸金属离子配合物的稳定性,结果表明,在气相和液相下都是氨基酸两性离子与金属离子的双齿配位体最稳定.
α-丙氨酸(α-Ala)是结构最简单的手性氨基酸,按构型分为S-α-Ala和R-α-Ala,根据旋光性分为左旋体(L-α-Ala)和右旋体(D-α-Ala),L-α-Ala在生命体内具有生物活性[7].α-Ala的手性特征使其金属配合物也具有手性,α-Ala金属配合物的研究对研究其它氨基酸金属配合物具有指导意义.
通常手性药分子只是一种对映体有药理活性,另一种对映体没用甚至有副作用.研究手性氨基酸金属配合物的旋光异构,对指导人们科学使用氨基酸金属盐进行氨基酸与金属离子的同补有重要意义.基于此,学者们对氨基酸与金属离子配合物的旋光异构进行了研究,文献[8-16]研究表明,气相下α-丙氨酸与Cu2+、Fe2+和Zn2+等配合物以及脯氨酸Ca2+配合物很难消旋,说明了这些氨基酸金属配合物在气固相可以安全地保存.文献[17-23]研究表明,水液相下α-Ala与Zn2+、Ca2+、Mg2+和Fe2+配合物以及脯氨酸与Ca2+配合物只能微量或极少量的消旋,说明α-丙氨酸二价锌(Zn)、钙(Ca)、镁(Mg)和铁(Fe)盐及脯氨酸二价钙盐用于生命体同补相关的氨基酸和金属离子具有较好的安全性.
钴是生物体必需的微量元素,它对氨基酸代谢、造血功能和甲状腺肿大的防治有重要作用,钴缺乏可诱发贫血和老年痴呆等疾病[24].生命体严重缺钴需用药物补钴,目前,药物补钴的途径主要是用无机钴盐.但无机钴盐补钴会导致生命体在短时间内钴含量迅速增加,对生命体产生毒副作用.基于氨基酸盐具有缓慢释放金属离子的优点,用氨基酸盐补钴,还可以达到补充氨基酸的目的,人们开始考虑使用氨基酸盐补钴.生命体需要的是二价钴(Co2+)和三价钴(Co3+),并且三价钴更重要[25].关于α-丙氨酸Co3+配合物旋光异构的研究未见报道,为揭示使用丙氨酸三价钴盐同补α-Ala与Co3+是否安全,结合文献[8-23]的研究经验,本工作对α-Ala·Co3+的旋光异构进行理论研究.
1 计算方法及模型选取
在M06[26]/6-311+G(d,p)水平优化气相反应过程单重态(计算表明本研究体系在单重态最稳定)势能面上的驻点结构(自由能热校正在1个标准大气压298.15 K温度下进行);采用SMD[27]模型方法,在SMD/M06/6-311+G(d,p)水平优化水液相下的驻点结构.通过对过渡态[28]内禀反应坐标(IRC)的计算[29],对过渡态的可靠性予以确认.为得到精确的反应势能面,在MN15[30]/6-311++G(2df,pd)水平计算驻点的单点能.总自由能是自由能热校正和单点能之和,势能曲线的零势点为该曲线上各个驻点的相对势能零点.采用NBO 5.0程序(Theoretical Chemistry Institute, University of Wisconsin, USA, 2001)中的自然键轨道(NBO)方法和AIM 2000程序(Version 2.0, McMaster University, USA, 2002)中的分子中的原子(AIM)方法,计算并分析了主要驻点物种的NPA电荷、成键临界点(BCP)、成环临界点(RCP)的电荷密度(ρ)以及电荷密度的拉普拉斯值(∇2ρ).气相下S-α-Ala与Co3+的配合物S-α-Ala·Co3+记作S-A·Co(Ⅲ),水相下各个驻点X记作X@W.S-A·Co(Ⅲ)在a、b和c通道异构的第1个公用的无手性过渡态记作T1a(b,c),中间体记作T1a(b,c);在a通道的手性对映体产物记作R-A·Co(Ⅲ)a.水相下4个H2O与T1a(b,c)的Co3+配位,2聚水与T1a(b,c)氢键作用的体系记作T1a(b,c)←4H2O·(H2O)2@W,其它体系表示法相似,络合物内的虚线表示分子间的氢键作用.文中计算工作采用Gaussian 16 Revision C.01程序.
2 结果与讨论
计算表明,气相和水液相下α-Ala·Co3+都是两性α-Ala分子的羧基O与Co3+二配位的双齿配合物构象最稳定,并且气相和水液相下双齿配合物α-Ala·Co3+的几何构象相差不明显.为节省篇幅,先给出气相下优化的α-Ala·Co3+双齿配合物的手性对映体几何构型(图1).
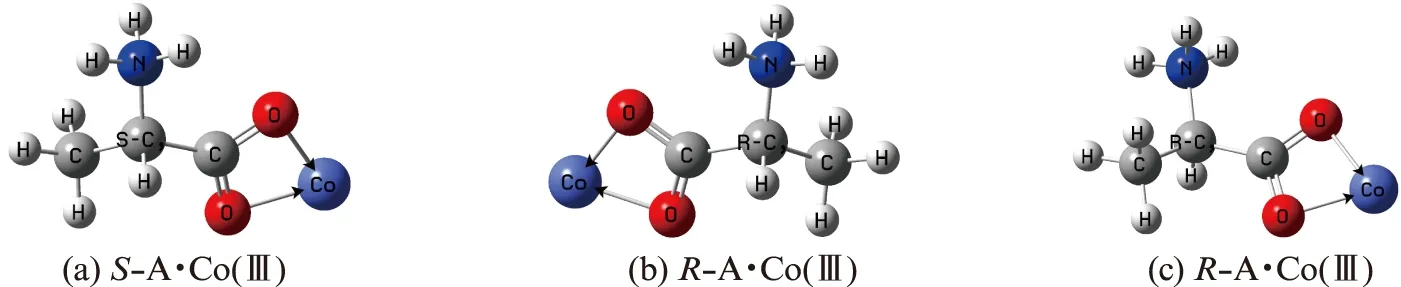
图1 α-Ala和Co3+配合物分子手性对映体的几何构型Fig.1 Geometric conformation of α-Ala and Co3+ complex molecule chiral enantiomer
对气相体系的研究,既可获得体系本质的物理和化学性质,揭示气相下体系的稳定性及变化规律,又可为进一步研究体系在其它复杂环境下的行为奠定基础[31-32],而生命体主要是水环境,因此,本工作先讨论S-A·Co(Ⅲ)分子在气相下的旋光异构,而后讨论其在水液相下的旋光异构.
2.1 气相S-A·Co(Ⅲ)的旋光异构
研究表明,气相S-A·Co(Ⅲ)可在质子以羰基O为桥以及羰基O和甲基C联合为桥迁移的2个通道a和b实现旋光异构,反应历程见图2,反应的自由能势能面见图3.气相S-A·Co(Ⅲ)在质子以羰基O和氨基N为桥以及质子先从质子化氨基向羰基O迁移的2个通道c和d上并没有旋光异构,而是解离成2个和3个碎片.这不同于气相S-A·Co(Ⅱ)以及气相下α-丙氨酸与Cu2+、Ca2+、Na+、Zn2+和Mg2+等配合物在这两个通道可以旋光异构[8-16].为了使读者全面了解气相S-A·Co(Ⅲ)的质子反应机理,本工作对a、b、c和d通道分别给予讨论.
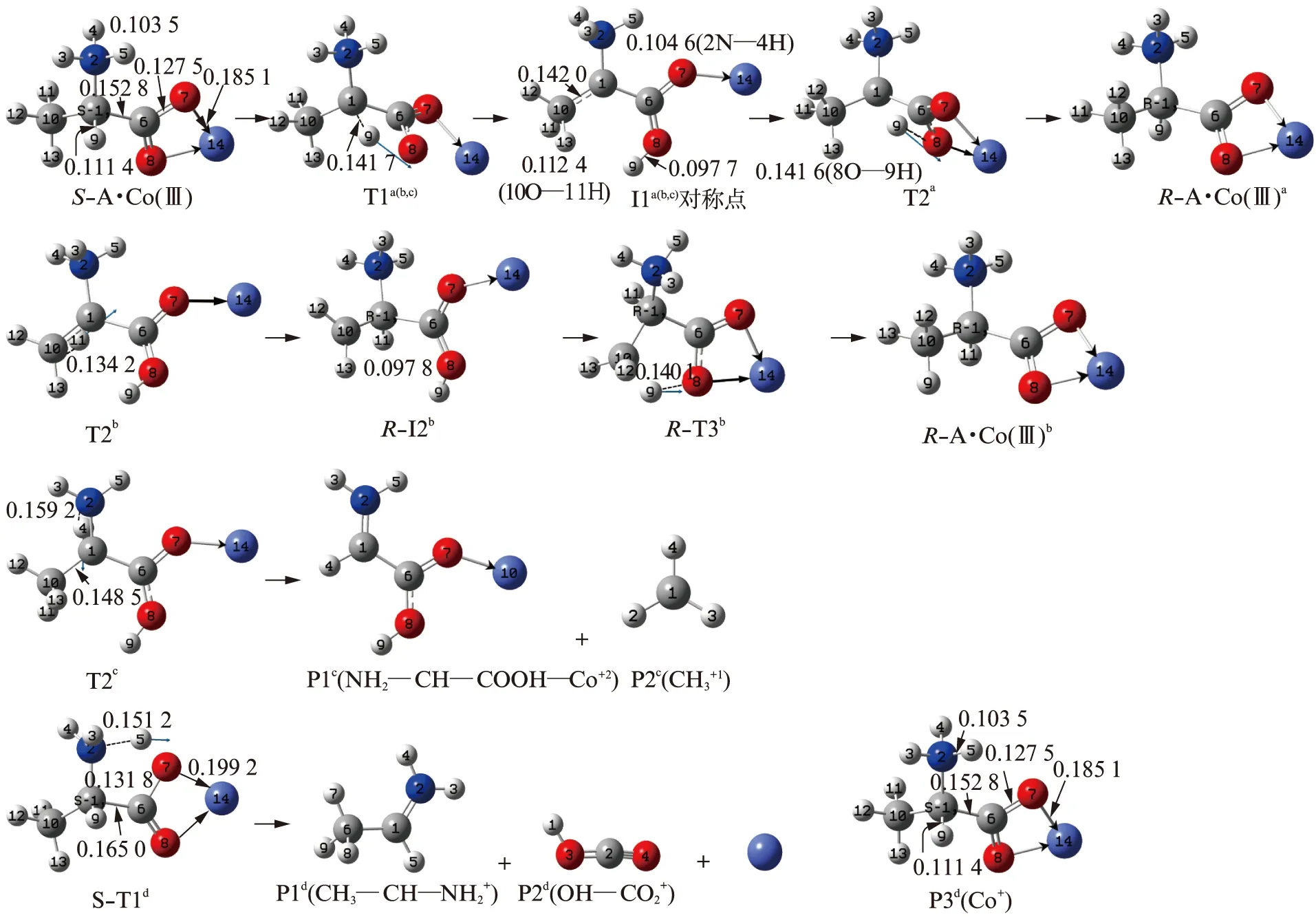
图2 气相S-A·Co(Ⅲ)旋光异构及解离的反应历程(键长单位: 纳米)Fig.2 Reaction process of optical isomerism and dissociation of S-A·Co(Ⅲ) in the gas phase(Bond length unit: nanometer)

图3 气相S-A·Co(Ⅲ)旋光异构及解离反应的自由能势能面Fig.3 The free energy potential surface of optical isomerism and dissociation of S-A·Co(Ⅲ) in the gas phase
a、b和c通道:
第1基元是a、b和c通道共用,十分重要,给予深入讨论.S-A·Co(Ⅲ)经过渡态T1a(b,c),9H从1C迁移到8O,异构成I1a(b,c).从S-A·Co(Ⅲ)到T1a(b,c),1C—9H从0.111 4 nm拉伸至0.141 7 nm,1C—9H的BCP电荷密度ρ从0.265 3 a.u.降到0.126 5 a.u.,拉普拉斯值(∇2ρ)始终为负值,1C—9H的共价键由强变弱;9H—8O的ρ从0.0 a.u.变为0.106 5 a.u.∇2ρ从0.0 a.u.变为正值,9H—8O从不成键到氢键作用;1C—9H—8O—6C的RCP电荷密度ρ从0.0 a.u.变为0.080 0 a.u.,∇2ρ从0.0 a.u.变为正值,从不成键到π键作用,环形过渡态形成;2N—1C的ρ从0.234 6 a.u.增加到0.249 5 a.u.,∇2ρ始终为负值,2N—1C的共价键增强;6C—1C和10C—1C两个共价键显著增强、螯合环6C—8O—14Cu—7O的π键作用减弱,8O—6C共价键稍有变弱(6C—8O从0.128 1 nm拉伸至0.129 8 nm)、配位键8O—14Cu和7O—14Cu稍有减弱(为节省篇幅ρ和∇2ρ的数据变化从略);二面角2N—1C—10C—6C从128.8°增加到155.4°;6C—1C键右视顺时针内旋转68.1°,这些变化使T1a(b,c)产生了113.2 kJ·mol-1的内禀能垒.该能垒远小于S-A·Co(Ⅱ)旋光异构此基元反应的能垒271.7 kJ·mol-1[17],原因有2个,一是S-A·Co(Ⅱ)的1C—9H键长为0.109 8 nm,红外振动频率是3 039.8 cm-1,而S-A·Co(Ⅲ)的1C—9H键长是0.111 4 nm,红外振动频率红移到2 875.6 cm-1,S-A·Co(Ⅲ)的1C—9H键活化程度高得多;二是从S-A·Co(Ⅲ)到T1a(b,c)比从S-A·Co(Ⅱ)到T1a(b)过程1C—9H键的拉伸幅度[17]小很多.
从图3势能面可以看出,I1a(b,c)处在势阱底部,比S -A·Co(Ⅲ)稳定许多.原因主要有3个,一是I1a(b,c)的骨架原子以及5H、9H和12H形成了超共轭大π键;原因之二是从S -A·Co(Ⅲ)到I1a(b,c),骨架原子间距1C—2N、1C—10C、1C—6C、6C—7O和6C—8O从0.150 9、0.150 8、0.152 8、0.127 5和0.128 1 nm分别缩小到0.145 6、0.142 0、0.149 5、0.125 1、0.127 1 nm,对应的共价键增强;三是8O—14Cu和7O—14Cu配位键增强(为节省篇幅,所有BCP电荷密度ρ的变化从略).
接下来的反应历程分为3个分通道a、b和c.
a通道专属的反应:
第2基元.I1a(b,c)经过渡态T2a,9H在纸面里从8O向1C迁移,异构成R-A·Co(Ⅲ)a,S-A·Co(Ⅲ)实现旋光异构.从I1a(b,c)到T2a,8O—9H键从0.097 7 nm拉伸至0.141 6 nm断裂;1C—2N、1C—10C、1C—6C、6C—7O和6C—8O分别从0.145 6、0.142 0、0.149 5和0.125 1 nm拉伸到0.148 6、0.147 0、0.150 4和0.129 1 nm;6C—1C右视逆时针内旋转76.7°,这些变化使T2a产生了314.0 kJ·mol-1的内禀能垒.该能垒如此高的主要原因有两个,一是I1a(b,c)的8O—9H键红外振动频率很大,达到了3 681.6 cm-1,8O—9H键钝化程度高,二是I1a(b,c)形变要克服体系共轭大π键的作用.
b通道专属的反应:
第2基元.I1a(b,c)经过渡态T2b,11H在纸面里从10C迁移到1C,异构成中间体R-I2b,S-A·Co(Ⅲ)在b通道实现旋光异构.从I1a(b,c)到T2b,10C—11H键从0.112 4 nm拉伸至0.134 2 nm断裂,T2b产生的能垒是20.1 kJ·mol-1.此过程能垒如此之小,原因之一是10C—11H键是0.112 4 nm,红外振动频率是2 784.4 cm-1,10C—11H键已经被较好的活化;原因之二从I1a(b,c)到T2b,质子逆着体系的偶极矩方向迁移,体系的电场力助力质子迁移.
第3基元.R-I2b经过渡态R-T3b,9H从8O迁移到10C,异构成产物R-A·Co(Ⅲ)b,结构分析表明,S-A·Co(Ⅲ)在b通道实现了手性对映体转变.从R-I2b到R-T3b,8O—9H键从0.097 8 nm拉伸至0.140 1 nm断裂,R-T3b产生的能垒是201.8 kJ·mol-1.此过程能垒很大,原因之一是8O—9H键的红外振动频率是3 663.8 cm-1,8O—9H键比较钝化;原因之二从R-I2b到R-T3b,质子是顺着偶极矩方向迁移,体系的电场力对质子迁移过程做负功.
c通道专属的反应:
第2基元.I1a(b,c)经过渡态T2c,4H在纸面内侧从2N迁移到1C,解离成2个碎片产物P1c(NH2—CH—COOH—Co+2)和P2c(CH3+1).从I1a(b,c)到T2c,2N—4H键从0.104 6拉伸至0.159 1 nm断裂,1C—10C键从0.142 0拉伸至0.148 5 nm,T2c产生的内禀能垒是257.8 kJ·mol-1.
d通道:
S-A·Co(Ⅲ)经过渡态S-T1d,5H从2N迁移到7O,解离成3个碎片产物P1d(CH3—CH—NH2+)、P2d(OH—CO2+)和P3d(Co+).从S-A·Co(Ⅲ)到S -T1d,2N—5H键从0.103 5拉伸至0.151 2 nm断裂,1C—6C键从0.152 8拉伸至0.165 0 nm,6C—7O键从0.127 5拉伸至0.131 8 nm,7O—14Co键从0.185 1拉伸至0.199 2 nm,这些变化使S -T1d产生了305.1 kJ·mol-1的内禀能垒.
从图3可以看出,S-A·Co(Ⅲ)在a和b通道旋光异构反应的活化能(气相反应势能面的表观能垒)是113.2 kJ·mol-1,来自于过渡态T1a(b,c);比较而言b通道比a通道具有优势.从图3还可以看出,S-A·Co(Ⅲ)在c通道的解离反应的活化能也是113.2 kJ·mol-1,在d通道的解离反应的活化能是305.1 kJ·mol-1.由于能垒113.2 kJ·mol-1高于温和反应能垒80.0 kJ·mol-1[33]不少,因此,S-A·Co(Ⅲ)在a和b通道的旋光异构以及在c通道的解离过程进行的比较缓慢.由于305.1 kJ·mol-1远高于极限反应能垒167.0 kJ·mol-1[33],因此S-A·Co(Ⅲ)在d通道的解离过程实际不可能发生.
2.2 水液相下S-A·Co(Ⅲ)的旋光异构
非氢转移反应的能垒较小,水分子(簇)与反应物作用(氢键作用及配位作用)对能垒影响不明显,考虑隐性水溶剂效应(亦即溶剂极性的作用)即可说明相关的反应机制[16,20].氢迁移反应能垒一般较高,水分子(簇)做氢转移媒介以及水分子与金属离子配位会显著改变反应能垒,研究氢迁移反应要同时考虑溶剂极性和水分子(簇)的共同作用,即显性水溶剂效应[14-23].为清晰地呈现水液相下S-A·Co(Ⅲ)旋光异构的历程以及水分子(簇)所起的作用,先讨论隐性水溶剂效应下S-A·Co(Ⅲ)的旋光异构,然后再讨论显性水溶剂效应下S-A·Co(Ⅲ)旋光异构过程中的氢迁移反应.
2.2.1 隐性水溶剂效应下S-A·Co(Ⅲ)的旋光异构
研究表明,水液相下S-A·Co(Ⅲ)的旋光异构可在气相下的a和b通道以及质子以羰基O和氨基N为桥迁移的c通道实现,反应历程见图4,反应的自由能势能面见图5(第612页).水液相下S-A·Co(Ⅲ)的质子化氨基上的质子不能从氨基N向羰基O迁移,这不同于水液相下S-A·Co(Ⅱ)以及α-丙氨酸与Cu2+、Ca2+、Na+、Fe2+、Zn2+和Mg2+等配合物旋光异构[8-16]的情况.
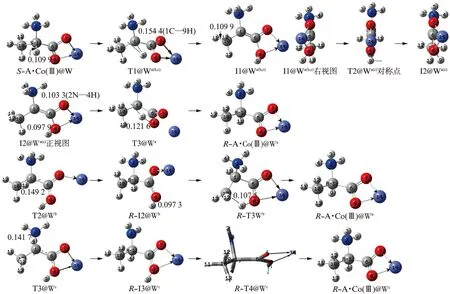
图4 隐性水溶剂效应下S-A·Co(Ⅲ)的旋光异构反应历程(键长单位: 纳米)Fig.4 Reaction process of S-A·Co(Ⅲ) optical isomerism under the effect of recessive water solvent(Bond length unit: nanometer)

图5 隐性水溶剂效应下S-A·Co(Ⅲ)的旋光异构反应的自由能势能面Fig.5 The free energy potential surface of S-A·Co(Ⅲ) optical isomerism reaction under the effect of recessive water solvent
a通道
第1基元,a、b和c通道共用.S-A·Co(Ⅲ)@W经过渡态T1@Wa(b,c),9H从1C迁移到8O,异构成I1@Wa(b,c).从S-A·Co(Ⅲ)@W到I1@Wa(b,c),1C—9H从0.109 9 nm拉伸至0.154 4 nm,1C—9H的BCP电荷密度ρ从0.276 4 a.u.降到0.098 5 a.u.,拉普拉斯值(∇2ρ)的符号由负变为值,1C—9H由共价键变为氢键;9H—8O的ρ从0.0 a.u.变为0.171 0 a.u.∇2ρ从0.0 a.u.变为负值,9H—8O从不成键到共价键作用;1C—9H—8O—6C的RCP电荷密度ρ从0.0 a.u.变为0.085 1 a.u.,∇2ρ从0.0 a.u.变为正值,从不成键到π键作用,环过渡态形成;2N—1C的ρ从0.242 7 a.u.增加到0.262 4 a.u.,∇2ρ始终为负值,2N—1C的共价键增强;6C—1C和10C—1C两个共价键显著增强、螯合环6C—8O—14Cu—7O的π键作用减弱、8O—6C共价键稍有变弱、配位键8O—14Cu和7O—14Cu稍有减弱(为节省篇幅ρ和∇2ρ的数据变化从略).二面角2N—1C—10C—6C从123.4°增加到149.6°;6C—1C键右视顺时针内旋转53.5°,这些变化使T1@Wa(b,c)产生了235.6 kJ·mol-1的内禀能垒.该能垒远大于T1a(b,c)的能垒113.2 kJ·mol-1,原因有3个,一是S-A·Co(Ⅲ)@W的1C—9H键长缩小到0.109 9 nm,红外振动频率蓝移到3 053.1 cm-1,S-A·Co(Ⅲ)@W的1C—9H键钝化了;二是从S-A·Co(Ⅲ)到T1a(b,c),质子迁移方向逆着体系的偶极矩矢量,体系的电场力助力质子迁移,而从S-A·Co(Ⅲ)@W到T1@Wa(b,c),质子迁移方向与偶极矩矢量垂直,体系电场力不对质子迁移做功;三是从S-A·Co(Ⅲ)到T1a(b,c),1C—9H的共价键由强变弱,并未断裂,而S-A·Co(Ⅲ)@W到T1@Wa(b,c),1C—9H由共价键变为氢键,共价键断裂.
第2基元,a和c通道共用.I1@Wa(b,c)经9H左右翻转的过渡态T2@Wa(c)(右视图),异构成I2@Wa(c).从I1@Wa(b,c)到T2@Wa(c),二面角9H—8O—6C—1C从-34.7°变为0.0°,8O—6C键仰视逆时针内旋转34.7°;二面角14Co—7O—6C—8O从-19.0°变为0.0°,7O—6C键俯视逆时针内旋转19.0°.2个化学键的内旋转所需能量很小,T2@Wa(c)产生的内禀能垒仅有3.3 kJ·mol-1.
第3基元,a通道专属的反应.
I2@Wa(c)经过渡态T3@Wa,9H在纸面里从8O迁移到1C,异构成产物R-A·Co(Ⅲ)@Wa,S-A·Co(Ⅲ)@W实现旋光异构.从I2@Wa(c)到T3@Wa,8O—9H键从0.097 9 nm拉伸至0.121 6 nm;6C—1C右视逆时针内旋转95.6°,这些变化使T3@Wa产生了175.3 kJ·mol-1的内禀能垒.该能垒比2.1节中气相下此基元的能垒大幅下降,原因是I2@Wa(c)的偶极矩(3.923 3 D)很小,极性溶剂水的作用使其变得不稳定,使其上升到势能面较高的位置.
b通道专属的反应:
第2基元.I1@Wa(b,c)经过渡态T2@Wb,11H在纸面内侧从10C迁移到1C,异构成R-I2@Wb,S-A·Co(Ⅲ)在b通道实现旋光异构.从I1@Wa(b,c)到T2@Wb,10C—11H键从0.109 9 nm拉伸至0.149 2 nm断裂,T2@Wb产生的内禀能垒是96.3 kJ·mol-1.该能垒较气相下此基元的能垒大幅增加,原因是水液相下10C—11H键缩短,红外振动频率蓝移到3 046.81 cm-1,10C—11H键钝化了.
第3基元.R-I2@Wb经过渡态R-T3@Wb,9H从8O迁移到10C,异构成产物R-A·Co(Ⅲ)@Wb,结构分析表明,S-A·Co(Ⅲ)@W在b通道实现了手性对映体转变.从R-I2@Wb到R-T3@Wb,8O—9H键从0.097 3 nm拉伸至0.107 5 nm,R-T3@Wb产生的内禀能垒是54.6 kJ·mol-1.此能垒较气相下此基元的能垒大幅下降,原因是R-I2@Wb到R-T3@Wb比R-I2b到R-T3b过程8O—9H键的拉伸幅度小得多.
c通道专属的反应:
第3基元.I2@Wa(c)经过渡态T3@Wc,4H在纸面内侧从2N迁移到1C,异构成R-I3@Wc,S-A·Co(Ⅲ)在c通道实现了旋光异构.从I2@Wa(c)到T3@Wc,2N—4H键从0.1033拉伸至0.141 7 nm断裂,T3@Wc产生的能垒是233.2 kJ·mol-1.该能垒小于2.1节中气相下对应的此基元的能垒,原因是水液相下2N—4H键的拉伸幅度有所减小.
第4基元.R-I3@Wc经6C—1C键内旋转的过渡态R-T4@Wc,6C—1C键右视逆时针内旋转与9H从8O向2N迁移协同进行,异构成产物R-A·Co(Ⅲ)@Wc.结构分析表明,S-A·Co(Ⅲ)@W在c通道实现了手性对映体转变.从R-I3@Wc到R-T4@Wc,6C—1C键右视逆时针内旋转94.2°,化学键内旋转所需能量很少,R-T4@Wc产生的内禀能垒只有9.3 kJ·mol-1.
从图5可以看出,隐性溶剂效应下S-A·Co(Ⅲ)在a、b和c通道旋光异构的活化能在233.2至235.6 kJ·mol-1之间,没有哪个通道具有明显地优势.考虑到b通道的第2和第3基元内禀能垒明显低于a和c通道的第3基元,也可以说比较而言b通道具有优势.233.2 kJ·mol-1高于极限反应能垒167.0 kJ·mol-1[33]许多,因此,只考虑隐性溶剂效应时α-丙氨酸Co(Ⅲ)配合物不能旋光异构.
2.2.2 显性溶剂效应下的氢迁移反应
从图5可以看出,水液相下S-A·Co(Ⅲ)在b通道旋光异构的第2和3基元(均为氢迁移反应)的内禀能垒分别是96.3和54.6 kJ·mol-1,在常温下就能进行.为节省篇幅,本节对这两个基元不进行讨论.只对a通道的H迁移反应以及c通道第3基元进行讨论.
水分子多以水簇的形式存在,显性水溶剂效应下,水分子簇与14Co形成配位键,同时水分子簇与S-A·Co(Ⅲ)的氢、氧和氮等氢键作用.以往的研究表明,水簇或单个水分子与金属离子配位对反应能垒的影响很小[22-23],2聚水和3聚水做质子转移媒介时质子转移的能垒相差很小[34-36],只与底物分子氢键作用但没参与反应的水分子(簇)对反应能垒影响较小[37].因此为使问题简便,本工作只讨论单个水分子与14Co配位,2聚水与S-A·Co(Ⅲ)的氢、氧和氮等氢键作用并做质子转移媒介的情况.
Co3+最多是6配位,在S-A·Co(Ⅲ)的Co3+已与2个O配位,再有4个H2O与Co配位,Co便“满配”,得到的配合物是S-A·Co(Ⅲ)←4H2O.再有2聚水与α-H和羰基氧8O氢键作用,同时和与Co3+配位的H2O氢键作用,形成有氢键网络的底物S-A·Co(Ⅲ)←4H2O·(H2O)2m@W(m表示2聚水在纸面外侧与S-A·Co(Ⅲ)氢键作用).相似的形成中间体I2a←4H2O·(H2O)2n@W和I2c←4H2O·(H2O)2n@W(n表示2聚水在纸面内).S-A·Co(Ⅲ)←4H2O·(H2O)2m@W在a通道旋光异构过程中的H迁移反应以及c通道第3基元I2c←4H2O·(H2O)2n@W的异构历程见图6(第614页),反应的自由能势能面见图7(第614页).

图6 显性水溶剂效应下S-A·Co(Ⅲ)旋光异构的主要H迁移历程Fig.6 The main H transfer reaction process of S-A·Co(Ⅲ) optical isomerism under the effect of explicit water solvent
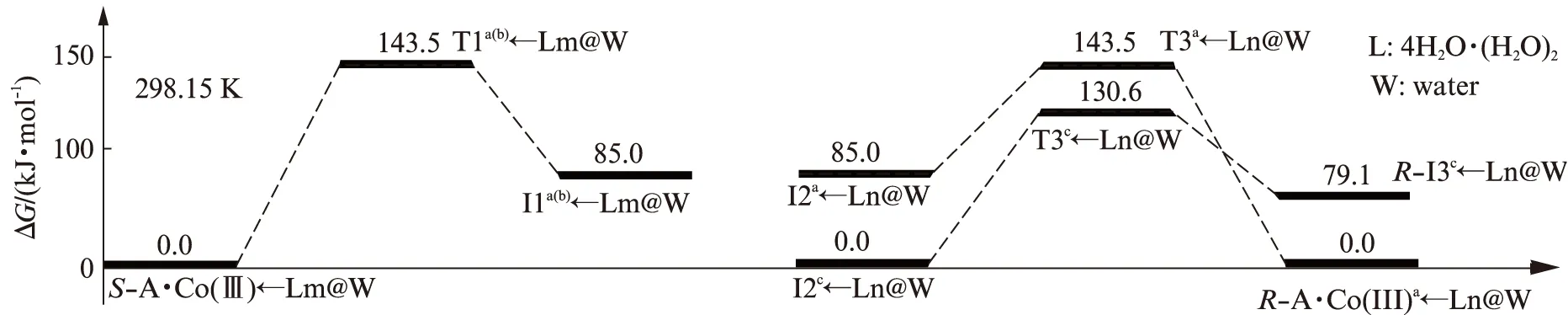
图7 显性水溶剂效应下S-A·Co(Ⅲ)旋光异构主要H迁移历程的自由能势能面Fig.7 Potential energy surface of free energy of main H transfer reaction process of S-A·Co(Ⅲ) optical isomerism under the effect of explicit water solvent
共用的第1基元,此基元十分重要,给予全面深入的讨论.
S-A·Co(Ⅲ)←4H2O·(H2O)2m@W经过渡态T1a(b,c)←4H2O·(H2O)2m@W,9H、29H和32H协同迁移,实现了质子从1C向8O净迁移,异构成中间体I1a(b,c)←4H2O·(H2O)2m@W.
从S-A·Co(Ⅲ)←4H2O·(H2O)2m@W到T1a(b,c)←4H2O·(H2O)2m@W过程,1C—9H、27O—29H和30O—32H键长从0.109 6、0.100 9和0.098 0 nm分别拉伸至0.151 9、0.109 2和0.098 7 nm,它们的BCP电荷密度(ρ)分别从0.278 5、0.292 5和0.336 7 a.u.减小到0.098 0、0.228 6和0.326 6 a.u.,∇2ρ始终为负值,1C—9H间的共价键显著变弱,27O—29H和30O—32H间的共价键有所变弱;8O—6C的BCP电荷密度(ρ)从0.341 1 a.u.减小到0.310 2 a.u.,∇2ρ始终为负值,共价键显著变弱;1C—9H—27O—29H—30O—32H—8O—6C的RCP电荷密度(ρ)从不存在到0.006 1 a.u.,∇2ρ为正值,八元环从没作用到π键作用,环形过渡态形成.二面角2N—1C—10C—6C从121.9°变为142.9°,这些变化使T1a(b,c)←4H2O·(H2O)2m@W产生了143.5 kJ·mol-1的内禀能垒.该能垒远远低于T1a(b,c)@W产生的内禀能垒235.6 kJ·mol-1,说明水分子(簇)对此基元反应起了很好的催化作用.原因是T1a(b,c)←4H2O·(H2O)2m@W的氢键角1C—9H—27O、27O—29H—30O和30O—32H—8O分别是165.7°、161.6°和158.1°,3个氢键较强,并且T1a(b,c)←4H2O·(H2O)2m@W的八元环结构共面程度较高,导致过渡态T1a(b,c)←4H2O·(H2O)2m@W有较好的稳定性.
a通道的第3基元.I2a←4H2O·(H2O)2n@W经过渡态T3a←4H2O·(H2O)2n@W,9H、31H和28H协同迁移,实现了质子在纸面里从8O向1C净迁移,得到R-型产物R-A·Co(Ⅲ)a←4H2O·(H2O)2n@W,S-A·Co(Ⅲ)←4H2O·(H2O)2m@W实现旋光异构.从I2a←4H2O·(H2O)2n@W到T3a←4H2O·(H2O)2n@W,8O—9H、30O—31H和27O—28H键从0.104 1、0.102 3和0.097 7 nm拉伸断裂,T3a←4H2O·(H2O)2n@W产生的内禀能垒是58.5 kJ·mol-1.该能垒远远低于前面T3a@W产生的内禀能垒175.3 kJ·mol-1,说明水分子(簇)起了显著的催化作用.原因有两个,一是T3a←4H2O·(H2O)2n@W的氢键角8O—9H—30O、30O—31H—27O和27O—28H—1C分别是158.1°、161.6°和165.7°,3个氢键较强,且八元环结构共面程度较好,导致T3a←4H2O·(H2O)2n@W的构象较稳定.二是从I2a←4H2O·(H2O)2n@W到T3a←4H2O·(H2O)2n@W是1C从sp2杂化向sp3杂化过渡,体系要释放些能量.
c通道的第3基元.I2c←4H2O·(H2O)2n@W经过渡态T3c←4H2O·(H2O)2n@W,实现质子在纸面里从2N向1C的净迁移,异构成R-I3c←4H2O·(H2O)2n@W,S-A·Co(Ⅲ)←4H2O·(H2O)2m@W在c通道实现旋光异构.从I2c←4H2O·(H2O)2n@W到T3c←4H2O·(H2O)2n@W,2N—4H、31O—30H和28O—27H键从0.108 3、0.100 5和0.097 5 nm分别拉伸至0.112 3、0.112 5和0.149 1 nm,T3c←4H2O·(H2O)2n@W产生的内禀能垒是130.6 kJ·mol-1.这远低于前面T3c@W产生的内禀能垒233.2 kJ·mol-1,水分子(簇)对此基元反应起了极显著的催化作用,原因相似于从I2a←4H2O·(H2O)2n@W到T3a←4H2O·(H2O)2n@W过程,不再赘述.
图7结合图5可以看出,水液相下S-A·Co(Ⅲ)在a、b和c通道旋光异构反应的活化能(决速步能垒)是143.5 kJ·mol-1,来自于第一基元反应的过渡态T1a(b,c)←4H2O·(H2O)2m@W.考虑到各个通道后续基元反应的能垒可知,比较而言S-A·Co(Ⅲ)在a通道的旋光异构更具优势.143.5 kJ·mol-1高于温和反应能垒80.0 kJ·mol-1[36]许多,比较接近极限反应能垒167.0 kJ·mol-1[36],说明水液相下α-丙氨酸三价钴的消旋反应只能极缓慢的进行,这从理论上说明α-丙氨酸三价钴盐用于生命体同补α-丙氨酸和三价钴离子具有很好的安全性.
3 结 论
采用M06和MN15方法,结合SMD模型方法,研究了α-Ala·Co3+的旋光异构及水溶剂效应,得到如下结果: 气相α-Ala·Co3+的旋光异构有2个通道,分别是羰基氧作质子迁移媒介和羰基氧和甲基碳联合作质子迁移媒介;水液相下α-Ala·Co3+可在3个通道实现旋光异构,与气相的情况相比,多了1个羰基氧和氨基氮联合作质子迁移媒介的反应通道.气相α-Ala·Co3+旋光异构的活化能(表观能垒)是113.2 kJ·mol-1,来自于质子从手性碳向羰基氧迁移的过渡态;水液相下α-Ala·Co3+旋光异构的活化能(决速步内禀能垒)是143.5 kJ·mol-1.
结果表明: 气相的α-Ala·Co3+可以缓慢地消旋,气相下α-Ala·Co3+不易保存;水液相下α-Ala·Co3+很难消旋,α-Ala·Co3+可以安全性用于生命体同补α-丙氨酸和三价钴离子.
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
云南化工(2021年8期)2021-12-21 06:37:38
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
质量技术监督研究(2018年1期)2018-03-26 08:04:28
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
光学精密工程(2016年2期)2016-11-07 09:02:32
