
人源β-N-乙酰葡糖胺转移酶I(hGnT-I)的原核表达与活性鉴定
2021-11-12 14:00陆天天高晓冬
食品与生物技术学报 2021年10期
陆天天,王 宁,高晓冬
(江南大学 生物工程学院,江苏 无锡 214122)
哺乳动物细胞中,蛋白质的N-糖基化修饰在内质网和高尔基体中完成,N-糖链结构依次经高甘露糖型、杂合型、最后生成成熟形态的复合型糖链,该过程对糖蛋白的结构稳定、半衰期和分子识别功能等具有关键作用[1]。β-N-乙酰葡糖胺转移酶I(GnT-I)是N-糖链加工过程中的重要蛋白质,它是一个定位在高尔基体上的膜蛋白,功能为以UDPGlcNAc为供体,向M5GN2寡糖的核心五糖结构中α1-3连接的甘露糖上添加一个 β1-2连接的GlcNAc[2-3],该步骤是N-糖链由高甘露糖型向杂合型或复合型转变的关键步骤[4-5]。
人源 GnT-I(hGnT-I)由 445 个氨基酸组成,相对分子质量约为50 900,1990年成功筛选到其基因MGAT1[2]。GnT-I蛋白已经成功在哺乳动物细胞[2]以及大肠杆菌中完成可溶性表达[6-7],目前市售GnT-I来自Novus Biologicals公司[8],为哺乳动物细胞体系表达的可溶性GnT-I蛋白。但哺乳动物细胞蛋白质表达量低,体系构建昂贵,导致该商业蛋白质产品价格很高,一定程度上阻碍了GnT-I的进一步研究和技术开发。原核表达体系虽然蛋白质产量高,但由于hGnT-I是带有一个跨膜域的膜蛋白,采用原核系统表达可能产生蛋白质降解、形成包涵体、所表达蛋白质无活性等问题。现有报道中的原核表达案例,均在GnT-I上融合了一段较长的蛋白质标签,如Seki等人报道在该蛋白质N端融合麦芽糖结合蛋白(MBP)以辅助其功能性表达与纯化[7],虽然得到了具有活性的蛋白质,但大部分蛋白质形成包涵体存在于沉淀里,大大降低了其应用性。
在本研究中,作者构建了用于表达截除N端跨膜域的重组hGnT-I的质粒,将其导入含有稀有密码子的大肠杆菌Rosetta DE3菌株中进行可溶性表达,随后优化了诱导表达条件并对纯化的蛋白质进行了活性研究。作者完成了重组hGnT-I大量功能性表达体系的建立,为进一步研究该蛋白质的性质及其在糖链合成中的应用打下了基础[9-10]。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 大肠杆菌菌种Rosetta DE3和表达载体pET28a质粒:购于德国Novagen公司;人基因组文库:购于美国Invitrogen公司;引物合成及基因测序:由华大基因完成。
1.1.2 培养基 LB培养基:酵母提取物5 g/L,胰蛋白胨 10 g/L,氯化钠10 g/L,琼脂粉 20 g/L(固体培养基),121℃灭菌20 min后备用;
TB培养基:酵母提取物24 g/L,胰蛋白胨12 g/L,磷酸氢二钾9.4 g/L,磷酸二氢钾2.2 g/L,甘油4 mL/L,121℃灭菌20 min后备用。
1.1.3 主要试剂 高速高保真PCR酶、限制性内切酶 BamH I和 Hind III、DNA Ligation Kit: 购于日本TaKaRa公司;异丙基-β-D-硫代半乳糖苷(Isopropyl-β-D-1-thiogalactopyranoside,IPTG):购于美国Sigma公司;氯霉素、卡那霉素:购于美国Gibco 公司;镍亲和小柱(HisTrap HP,1 mL):购于美国GE Healthcare公司;免疫印迹检测所用抗体Anti-His Mouse IgG和 Goat Anti-Mouse HRP:购于碧云天生物技术;荧光标记的糖链M3GN2-Asn-Fmoc和GN2M3GN2-Asn-Fmoc:获赠于日本产业技术综合研究所(AIST);核苷酸单糖 UDP-GlcNAc:购于青岛曙格公司;β-N-乙酰葡糖胺糖苷酶 (β-N-actylglucosiminidase S):购于美国NEB公司;其他常用试剂均购于中国国药集团。
1.1.4 主要仪器 高效液相色谱仪 (Waters 2695)和荧光检测器(Waters 2475):美国沃特世公司;色谱柱(Amide,150 mm×4.6 mm,3 μm):安捷伦科技有限公司;蛋白质电泳以及免疫印迹转移槽:美国伯乐公司。
1.2 方法
1.2.1 MGAT1基因的克隆及重组质粒pET28a-MGAT1ΔTM的构建 根据Genebank数据库人源MGAT1基因编码序列,结合UniPort数据库对蛋白质GnT-I跨膜区域的预测,人源MGAT1基因编码序列长1 335 bp,前87 bp编码一段面向胞质的片段以及跨膜域,故从第88 bp开始设计PCR引物,并在5′端分别引入BamH I和Hind III酶切位点,引物序列见表1。以人基因组文库中包含MGAT1基因的单克隆质粒为模板进行扩增,产物经1 g/dL琼脂糖凝胶电泳鉴定并进行PCR产物纯化,得到目的基因片段。将目的基因和pET28a空载质粒分别用BamH I和Hind III进行双酶切,产物经1 g/dL琼脂糖凝胶回收后以20 ng目的基因、10 ng pET28a、5 μL Ligation Mix体系在25℃连接15 min后将连接产物转化大肠杆菌DH5α,经SOC培养基在37℃预培养45 min后,涂布于LB/Kan+/Cm+(卡那霉素50 μg/mL,氯霉素 34 μg/mL)平板上进行筛选。 12 h后从平板挑取单克隆进行菌落PCR验证,将检定阳性的单克隆扩增并且提取质粒,命名为pET28a-MGAT1ΔTM。

表1 本研究所用的引物Table 1 Primers used in the study
1.2.2 重组hGnT-I蛋白的诱导表达 将pET28a-MGAT1ΔTM重组质粒转化入大肠杆菌Rosetta DE3感受态细胞中,涂布于LB/Kan+/Cm+平板上,于37℃培养过夜,挑取单菌落接种于5 mL LB/Kan+/Cm+(卡那霉素 50 μg/mL,氯霉素 34 μg/mL) 液体培养基中,于37℃培养12 h;后将培养基以1∶100体积比接入200 mL TB/Kan+/Cm+(抗生素质量浓度同上)液体培养基中,于37℃培养至OD600为0.8时,将培养基转移至16℃摇床冷却1 h,加入终浓度为100 μmol/L的IPTG,16℃诱导表达12 h。表达完成后于4℃、9 000 g离心3 min,收集菌体,弃尽培养液后于-80℃冰箱保存备用。
1.2.3 重组hGnT-I蛋白表达条件的优化 以1.2.2重组蛋白质的诱导表达为背景对蛋白质的表达条件进行优化。当重组菌扩大培养至OD600达到0.8时,降温至16℃后,加入 100 μmol/L的 IPTG,分别在 0、2、4、6、8、10、12、16 h 对菌液进行取样,采用免疫印迹法对不同诱导时间的蛋白质表达水平进行检测;采用相同的方法表达蛋白质,加入不同浓度的 IPTG(10、50、100、200、500、1 000 μmol/L),培养10 h后对菌液进行取样,采用免疫印迹法对加入不同浓度诱导剂的样品进行蛋白质表达水平检测。
1.2.4 重组hGnT-I蛋白的纯化 将冷冻保存的菌体置于冰水中解冻15 min,加入15 mL的重悬缓冲液 (25 mmol/L Tris-HCl,300 mmol/L NaCl,pH 8.0)重悬菌体,超声波破碎菌体10 min,于4℃、9 000 g离心45 min,收集上清液蛋白质。将上清液用注射器上样至用重悬缓冲液平衡的镍亲和层析小柱,并用含不同浓度咪唑(20、60、250 mmol/L)的重悬缓冲液洗脱,收集洗脱液,检测目的蛋白质。用30 000超滤离心管过滤含有重组hGnT-I(His-hGnT-IΔTM)蛋白的洗脱液,并加入25 mmol/L HEPES-NaOH(pH 7.4)洗涤3次,得到浓缩蛋白质液,置于-80℃冰箱备用。
1.2.5 重组hGnT-I蛋白的活性检测 糖基化反应(总体积 50 μL) 各组分如下:1 μmol/L M5GN2-Asn-Fmoc,0.1 mmol/L UDP-GlcNAc,10 mmol/L MgCl2,0.1 mg/mL His-hGnT-IΔTM 蛋白,50 mmol/L MES-NaOH(pH 6.0)。反应体系在37℃下孵育1 h,18 000 g离心5 min,取上清液进行HPLC检测。流动相 A:乙腈;流动相 B:0.1 mol/L NH4OAc。梯度:0~5 min,85%B;5~35 min,85%~50%B;35~40 min,50%~30%B;40~45 min,30%~85%B。 荧光检测器激发波长(ex):295 nm;发射波长(em):315 nm;流量:1.0 mL/min;柱温 40 ℃。
1.2.6 重组hGnT-I蛋白的底物特异性检测 底物特异性检测时,除将底物更换为M3GN2-Asn-Fmoc外[11],使用的反应条件和检测方法与活性检测相同,其中考虑到非天然底物的特异性问题,可以适当延长反应孵育时间。
1.2.7 产物的酶切验证 对1.2.5和1.2.6中的反应体系进行产物验证,方法如下:50 μL糖基化反应体系于100℃加热5 min,取44 μL与β-N-乙酰氨基葡糖苷酶[12](1 μL)和 10×GlycoBuffer(5 μL)混合,37℃孵育12 h,18 000 g离心5 min,取上清液用HPLC检测产物。
2 结果与分析
2.1 hGnT-I蛋白跨膜域结构分析
TMHMM跨膜域预测图谱见图1。可以看出,hGnT-I在N端有一个跨膜域结构,结合UniPort蛋白质数据库的模拟预测可知,跨膜区域位于7~29位氨基酸,而1~6位氨基酸位于胞质侧。本研究设计截除前87 bp编码1~29位氨基酸的序列,只表达包含高尔基体腔内侧可溶性片段的hGnT-IΔTM。
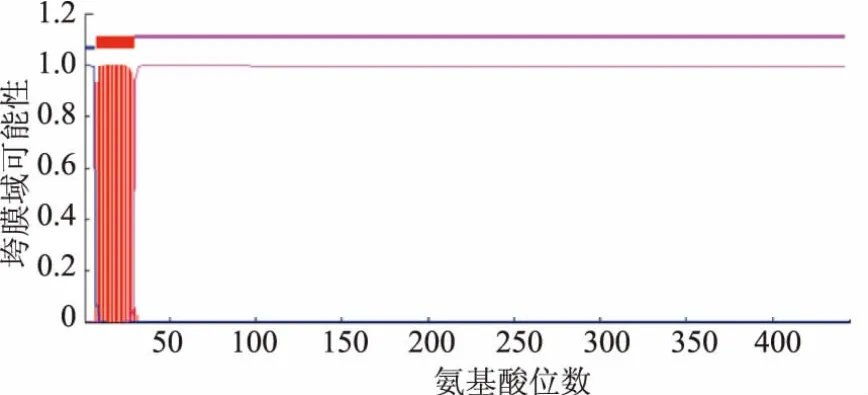
图1 hGnT-I蛋白的跨膜结构域预测Fig.1 Predicted transmembrane domain of hGnT-I
2.2 重组质粒pET28a-MGAT1ΔTM的构建
表达质粒选用多克隆区域前后都有6×His标签的原核表达载体pET28a,在蛋白质N端融合了一个6×His标签以进行后续纯化工作,重组质粒pET28a-MGAT1ΔTM 见图2。
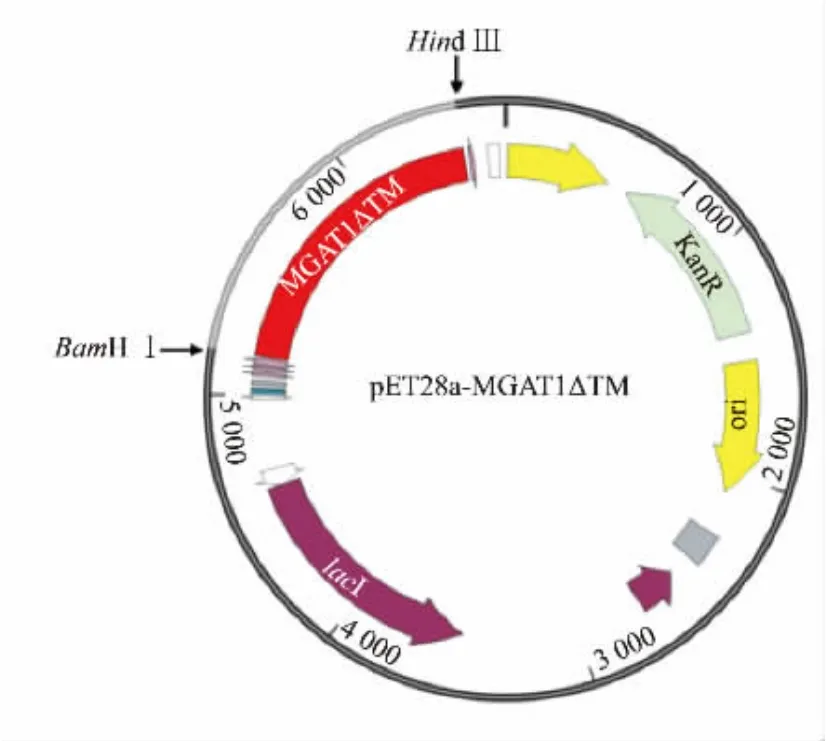
图2 重组质粒pET28a-MGAT1ΔTMFig.2 Recombinant expression plasmid pET28a-MGAT1ΔTM
2.2.1 基因的扩增 PCR产物经1 g/dL琼脂糖凝胶电泳分析,13 00 bp左右有明显条带,而截除前87 bp的目的基因MGAT1ΔTM片段长度为1 248 bp,与预期大小一致,见图3。
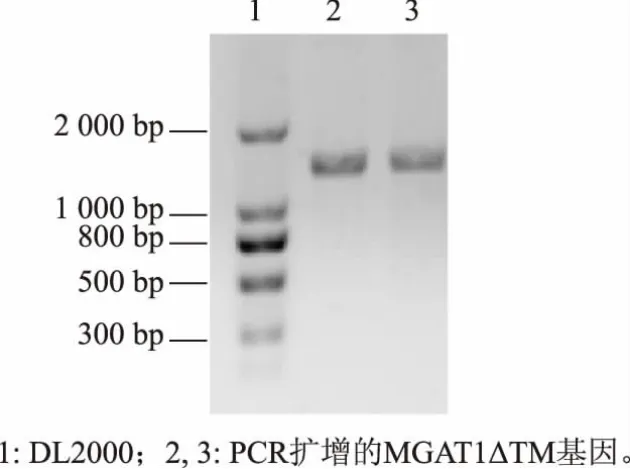
图3 MGAT1ΔTM目的基因PCR扩增结果电泳Fig.3 MGAT1ΔTM gene PCR amplification
2.2.2 重组质粒pET28a-MGAT1ΔTM菌落PCR验证及测序 经菌落PCR验证,所得质粒中至少3个单克隆扩增出1 300 bp左右的目的片段,见图4。挑取该3个阳性菌落进行扩增与质粒提取和测序,测序结果与Genbank中去除前87 bp的MGAT1基因编码区域一致,表明重组质粒pET28a-MGAT1ΔTM构建成功。
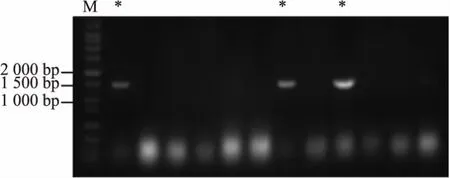
图4 pET28a-MGAT1ΔTM重组质粒菌落PCR验证Fig.4 Colony PCR for pET28a-MGAT1ΔTM plasmid construction
2.3 His-hGnT-IΔTM蛋白的表达与纯化
2.3.1 重组蛋白质的诱导表达和纯化 构建的重组质粒pET28a-MGAT1ΔTM转化入大肠杆菌Rosetta DE3中诱导12 h后,收集菌体进行超声波破碎,离心得到上清液;将上清液用镍亲和层析柱纯化,收集含不同浓度咪唑(20、60、250 mmol/L)缓冲液的洗脱液。对不同蛋白质样品进行SDS-PAGE凝胶电泳分析,结果见图5。与未加入IPTG诱导的细胞裂解液相比,诱导后的样品在42 800左右位置出现诱导前样品未出现的条带,表明重组hGnT-I蛋白可溶性表达成功,相对分子质量约为42 800,产率为7.5 g/L,见图5。
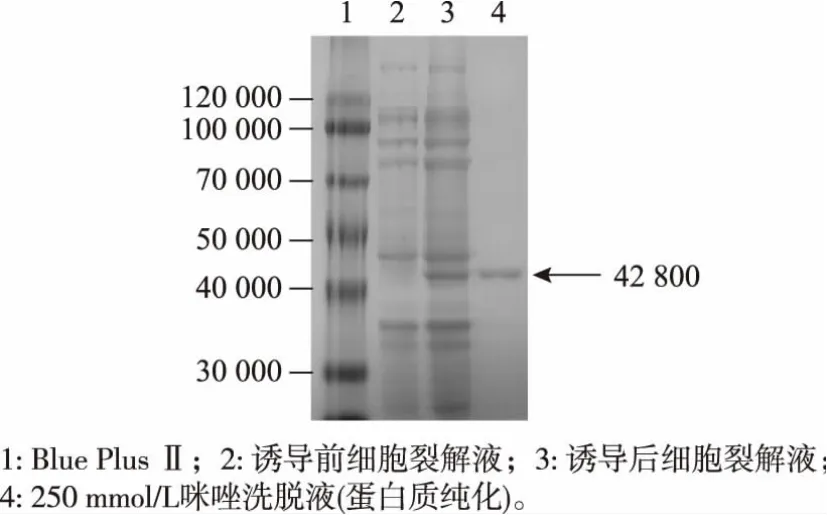
图5 重组蛋白His-hGnT-IΔTM的表达与纯化Fig.5 Expression and purification of recombinant HishGnT-IΔTM protein
经过SDS-PAGE及考马斯亮蓝染色验证,目的蛋白质集中于含250 mmol/L咪唑的洗脱液中。将该部分洗脱液收集并用超滤离心管过滤浓缩,通过反复用HEPES-NaOH缓冲液清洗后得到终质量浓度为5.0 mg/mL的重组hGnT-I蛋白。
2.3.2 重组蛋白质表达条件的优化 收集不同诱导条件下的菌体进行破碎裂解,直接将细胞裂解液作为样品进行免疫印迹法检测蛋白质表达程度,其中第一抗体为Mouse Anti-His IgG,第二抗体为Goat Anti-mouse IgG,HRP。结果表明,在诱导开始2 h后,蛋白质开始逐渐表达,直到10 h后表达量基本趋于一致,12 h后蛋白质表达量不再变化;采用优化过诱导时间10 h的进一步对加入的诱导剂浓度进行优化,结果表明,加入100 μmol/L IPTG比加入50 μmol/L可以使蛋白质表达量成倍增加,然而加入更高浓度的诱导剂并不能提高蛋白质的表达量,见图6。
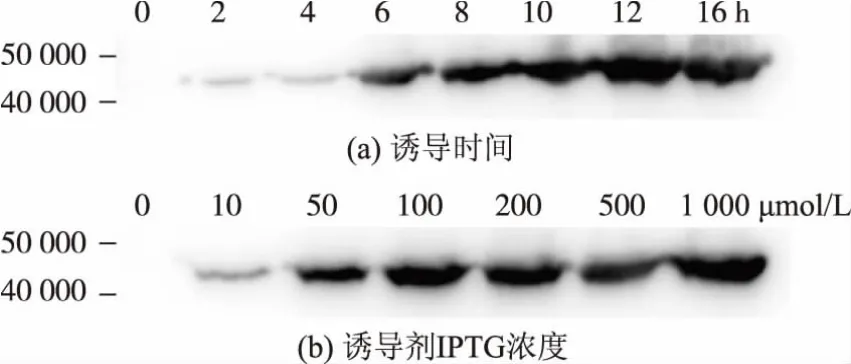
图6 重组hGnT-I蛋白表达条件的优化Fig.6 Optimization of the expression condition of hGnT-I protein
2.4 重组hGnT-I蛋白的性质鉴定
首先采用与天然底物类似的带荧光标签的N-糖链M5GN2-Asn-Fmoc作为重组hGnT-I蛋白的底物,确认了其体外活性,见图7。Stanly等人也于1990年报道hGnT-I蛋白在体外可以识别M3GN2,因此也检测了重组蛋白对非天然底物M3GN2-Asn-Fmoc的特异性,确认其可以作为重组hGnT-I的反应底物,见图8。反应产物经酶切验证为目标产物。
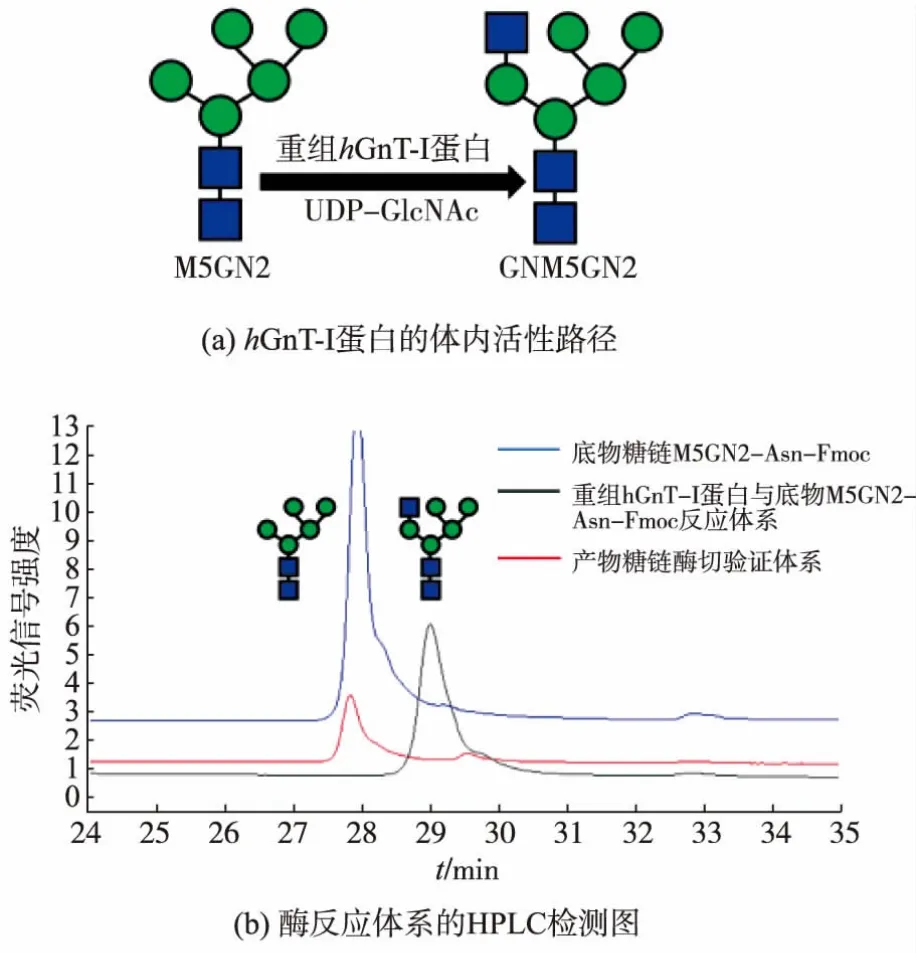
图7 重组hGnT-I蛋白对底物M5GN2-Asn-Fmoc的活性检测Fig.7 hGnT-I protein activity against M5GN2-Asn-Fmoc
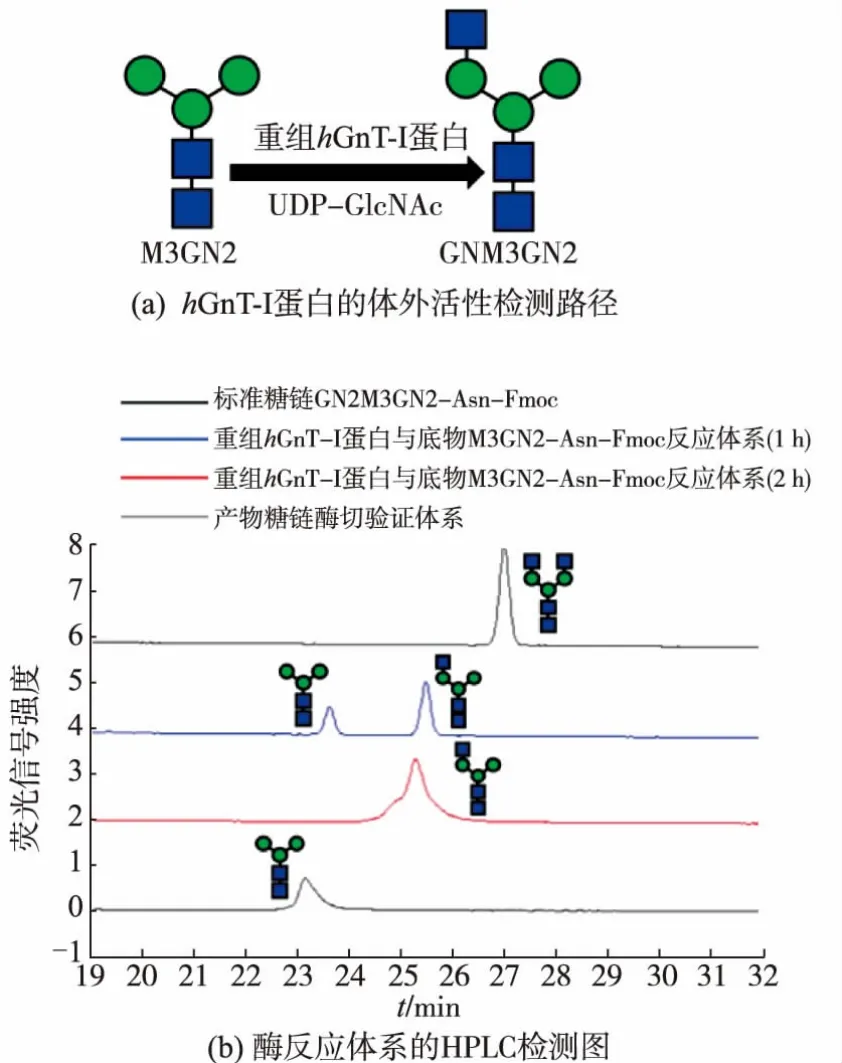
图8 重组hGnT-I蛋白对底物M3GN2-Asn-Fmoc的活性检测Fig.8 hGnT-I protein activity against M3GN2-Asn-Fmoc
2.4.1 重组蛋白质活性检测 以M5GN2-Asn-Fmoc为底物,体外糖基化反应体系离心取上清液后进行HPLC检测,结果见图7。反应体系中推测的糖链产物GNM5GN2-Asn-Fmoc较起始底物M5GN2-Asn-Fmoc有明显的偏移,间隔时间约1.2 min。说明重组hGnT-I蛋白成功以UDP-GlcNAc为糖基供体、M5GN2-Asn-Fmoc为受体进行了糖基化反应,生成了加上1个GlcNAc的产物糖链GNM5GN2-Asn-Fmoc,反应转化率为100%。
2.4.2 重组蛋白底物特异性检测 以M3GN2-Asn-Fmoc为底物经过1 h或2 h反应,从图8中可以看到,1 h反应体系中除了底物糖链M3GN2-Asn-Fmoc以外,还出现了添加了1个GlcNAc的产物GNM3GN2-Asn-Fmoc,而在延长孵育时间至2 h的样品中则只能检测到产物GNM3GN2-Asn-Fmoc。与标准糖链GN2M3GN2-Asn-Fmoc相比,该产物在HPLC中的洗脱时间较短,符合氨基柱以化合物极性大小正向洗脱的顺序,说明重组hGnT-I蛋白可以识别非天然底物M3GN2-Asn-Fmoc,但该酶促反应的效率低于天然底物。
2.4.3 反应产物的酶切验证 将反应体系中蛋白质高温灭活后,加入β-N-乙酰葡糖胺糖苷酶,经过12 h水解后对水解产物进行HPLC检测。结果显示,糖链产物GNM5GN2-Asn-Fmoc和GNM3GN2-Asn-Fmoc在糖苷酶的水解作用下,形成了未加上GlcNAc的底物M5GN2-Asn-Fmoc和M3GN2-Asn-Fmoc。该结果证明重组hGnT-I蛋白催化的糖基化反应为向寡糖底物添加β糖苷键连接的N-乙酰葡糖胺。
3 结 语
作者成功构建了重组质粒pET28a-MGAT1ΔTM,利用大肠杆菌表达体系实现了截除跨膜域的hGnT-I的大量可溶性表达,并在镍柱纯化后对该蛋白质进行了体外活性测试。研究结果表明,该重组蛋白质对天然底物类似物M5GN2-Asn-Fmoc和非天然底物M3GN2-Asn-Fmoc均具有活性。通过对反应产物进行酶切验证,证明糖基化产物为一个由β糖苷键连接的N-乙酰葡糖胺,说明该重组蛋白质的体外活性与体内活性一致。本研究克服了人源膜蛋白在原核体系表达易降解、易失活的难点,提供了大量获得活性重组hGnT-I蛋白的方法,为进一步研究其酶学性质以及糖链酶法合成相关技术提供了新的方法。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
粉末冶金技术(2021年3期)2021-07-28
江西农业学报(2021年4期)2021-04-20
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
三农资讯半月报(2020年11期)2020-06-21
天然产物研究与开发(2019年10期)2019-11-05
科技资讯(2018年16期)2018-10-26
今日财富(2017年32期)2017-10-19
