
口蹄疫病毒O 型泛亚株VP1 蛋白的二级结构及抗原表位预测
2021-11-11 03:56王国华田普厚李成龙刘冬冬张七斤
中国动物检疫 2021年11期
李 阳,马 园,王国华,田普厚,李成龙,刘冬冬,张七斤
(1.内蒙古农业大学兽医学院,内蒙古呼和浩特 010018;2.内蒙古必威安泰生物科技有限公司,内蒙古呼和浩特 011500)
口蹄疫(foot and mouth disease,FMD)是由口蹄疫病毒(foot and mouth disease virus,FMDV)感染牛、猪、羊等偶蹄动物引起的,具有高度传染性和重大经济意义的疫病,为世界动物卫生组织(OIE)要求的须通报动物疫病[1]。FMDV隶属于小RNA 病毒科口蹄疫病毒属(Aphthvirus),在免疫学上分为O、A、C、SAT1、SAT2、SAT3和Asia1 等7 种血清型[2]。FMDV 基因组全长约8 500 bp,由5'端非编码区、开放性阅读框(ORF)和3'端非编码区组成[3]。ORF(全长6 500 nt)编码4 种结构蛋白(VP1、VP2、VP3、VP4)和8 种非结构蛋白(Lpro、2A、2B、2C、3A、3B、3Cpro和3Dpol)。这些蛋白的主要功能是参加宿主免疫应答的产生和调节。其中VP1 蛋白暴露于病毒表面,最易发生变异,可使病毒突变株逃避抗体作用,从而获得持续感染机会[4]。FMDV 抗原位点多位于衣壳蛋白的环结构上,其中O 型FMDV至少包含5 个中和性抗原位点。O 型FMDV VP1蛋白作为ORF 区中P1 结构蛋白基因所编码的最重要的衣壳蛋白,在其GH 环(141~160 位氨基酸)和C 末端(200~213 位氨基酸)存在2 个线性表位,是FMDV 最重要的抗原位点,也是FMDV 变异的关键所在[5]。因此,VP1 蛋白是决定FMDV 抗原性诱导中和抗体和激发保护性免疫应答的主要抗原蛋白[6]。
近年来我国FMD 发病情况整体呈下降趋势,其发生主要与动物及其产品违规或跨境调运,导致新毒株输入有关,其中威胁最大的是来自东南亚、南亚和西亚-中东等地区的O、A 和Asia1 型[7]。例如:源自于南亚次大陆的O 型印度 2001 毒株(O/Ind-2001),2017 年由境外传入我国,现有 a、b、c、d、e 5 个进化分支[8]。源自东南亚的A 型东南亚 97 毒株(A/Sea-97)G1 和 G2 分支,分别于2009 年和2013 年传入我国。O 型缅甸 98 毒株(O/Mya-98),于2010 年传入我国[8]。而源自南亚的O 型泛亚(PanAsia)毒株早在1999 年前后就曾传入我国,2011 年再次经东南亚传入,开始在国内流行[9],成为当前和今后我国主要的流行毒株之一,须重点关注和防范[7],加强该毒株疫苗的研发。本研究分析O 型PanAsia 毒株VP1 蛋白的二级结构并预测其抗原表位,以期为该亚型毒株多表位疫苗的研制提供理论基础。
1 材料与方法
1.1 目的基因来源及编码信息
在GeneBank 上获取O/PanAsia 病毒株VP1基因序列(登录号MH807443.1,片段长度639 bp)。该基因编码的VP1 蛋白氨基酸序列如下:TTSTGESADPVTATVENYGGETQVQRRQHTDVS FILDRFVKVTPQNQINVLDLMQTPAHTLVGALLR TATYYFADLEVAVKHEGNLTWVPNGAPEAALDN TTNPTAYHKAPLTRLALPTAPHRVLATVYNGNCR YGTSPVTNVRGDLQVLAQKAARTLPTSFNYGAI KATRVTELLYRKRAETYCPRPLLAIHPSEARHKQ KIVAPVKQLL。
1.2 方法
1.2.1 理化性质分析 通过在线软件网站ProParam(https://www.expasy.org/resources/protparam),利用Expasy 工具分析其基本物理和化学性质[10]。
1.2.2 跨膜结构域、亲疏水性和信号肽预测 蛋白跨膜预测,使用TMHMM Server v.2.0(http://www.cbs.dtu.dk/services/TMHMM/)工具;蛋白信号肽预测,采用SignaIP 4.1 Server(http://www.cbs.dtu.dk/serv-ices/SignalP/)在线工具;蛋白亲疏水性预测,使用Expasy 网站的ProScale(https://web.expasy.org/protscale/)工具[11]。
1.2.3 二级结构预测 使用DNAstar Protean 和SOPMA(https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html)在线分析软件[12]。
1.2.4 三级结构预测 三级结构分析建模,采用SWISS MODE(https://www.expasy.org/resources/swiss-model)工具;三级结构预测,采用Phyre(http://www.sbg.bio.ic.ac.uk/phyre2/)软件[13]。
1.2.5 B 细胞表位预测 采用ABCpred(https://webs.iiitd.edu.in/raghava/abcpred/)软件和IEDB Analysis Resource(http://www.tools.iedb.org/main/)在线工具[14]。
1.2.6 T 细胞表位预测 利用SYFPEITHI(http://www.syfpeithi.de//bin/MHCServer.d11/)在线分析软件预测VP1 蛋白T 细胞表位;利用IEDB Analysis Resource(http://www.tools.iedb.org/main/)在线工具预测T 细胞抗原表位[15]。二者都是T 细胞受体(T-cell receptor/TCR)识别的抗原表位,但是预测方法不同,为后续结果提供了保障。
2 结果与分析
2.1 理化性质
根据ProParam 分析可知,VP1 蛋白由213 个氨基酸组成,其中占比最高的前3 位分别为苏氨酸(T,12.2%)、丙氨酸(A,11.3%)和亮氨酸(L,10.3%)。该蛋白相对分子质量为23 520.83,pI 值为9.49,负电荷残基总数(Asp+Glu)为16 个,正电荷残基总数(Arg+Lys)为23 个。分子式为C1046H1670N300O309S4,脂溶指数为86.57,不稳定性指数(II)为25.14,分类为稳定蛋白(>40 判定为不稳定蛋白);与FMDV O 型Ind-2001 毒株VP1 蛋白相比,O/PanAsia 毒株的VP1 蛋白相对分子质量略低,pI 值略高,虽然不稳定指数对比较高,但二者同为稳定蛋白。
2.2 亲疏水性、跨膜结构域和信号肽预测
由ProScale 软件分析可知,VP1 蛋白的平均亲水性系数(GRAVY)为-0.309,最大值为1.656,最小值为-2.567(图1),表明该蛋白为亲水蛋白(GRAVY 值范围为-2~2,负值表明为亲水性蛋白),比典型O 型Ind-2001 毒株VP1 蛋白(GRAVY 值为-0.327)低一些。
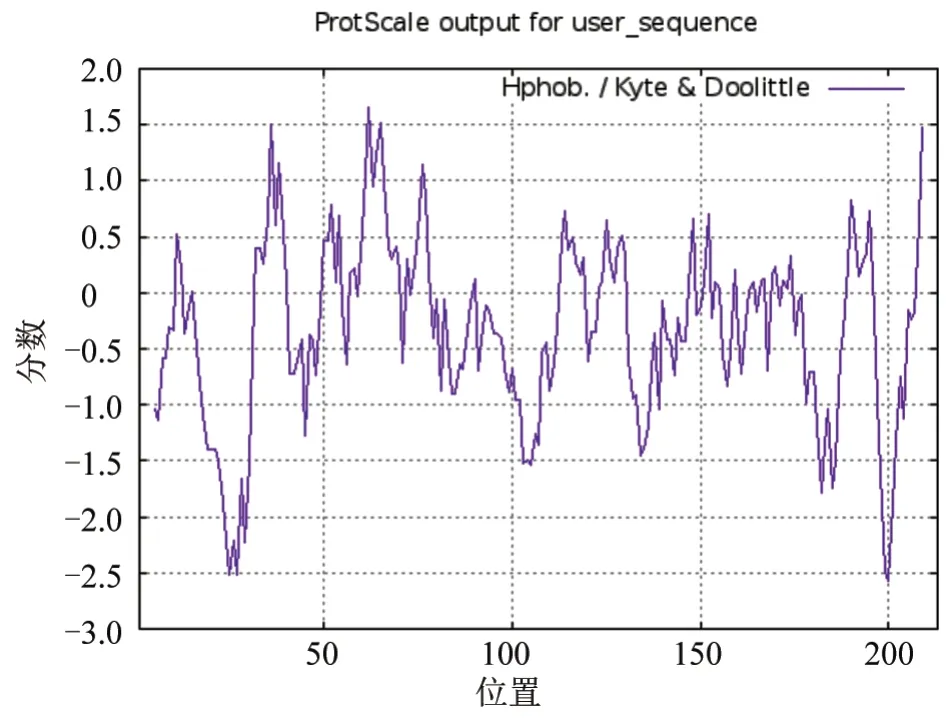
图1 VP1 蛋白亲疏水性分析结果
由TMHMM 软件分析可知,VP1 蛋白完全位于膜外,不存在跨膜结构域(图2);采用SignaIP 软件分析VP1 蛋白信号肽,发现该蛋白无信号肽存在(图3),表明其为非分泌型蛋白。
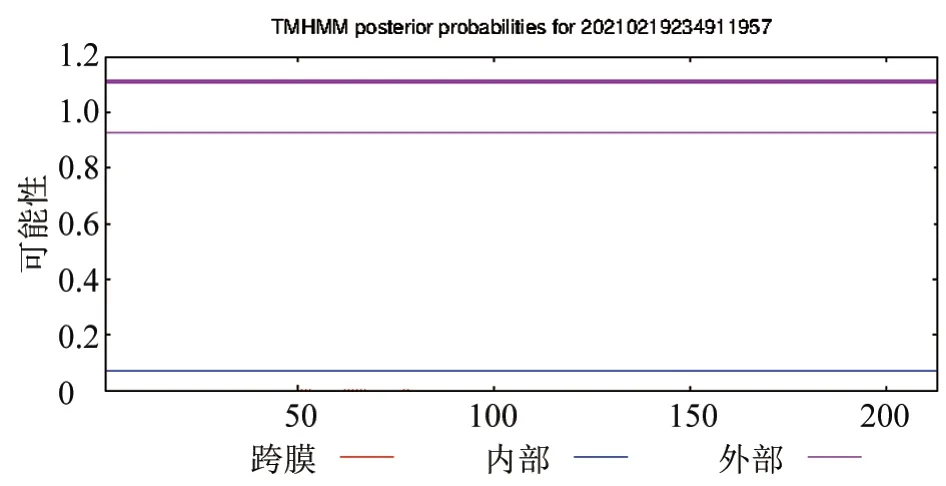
图2 VP1 蛋白跨膜区分析结果
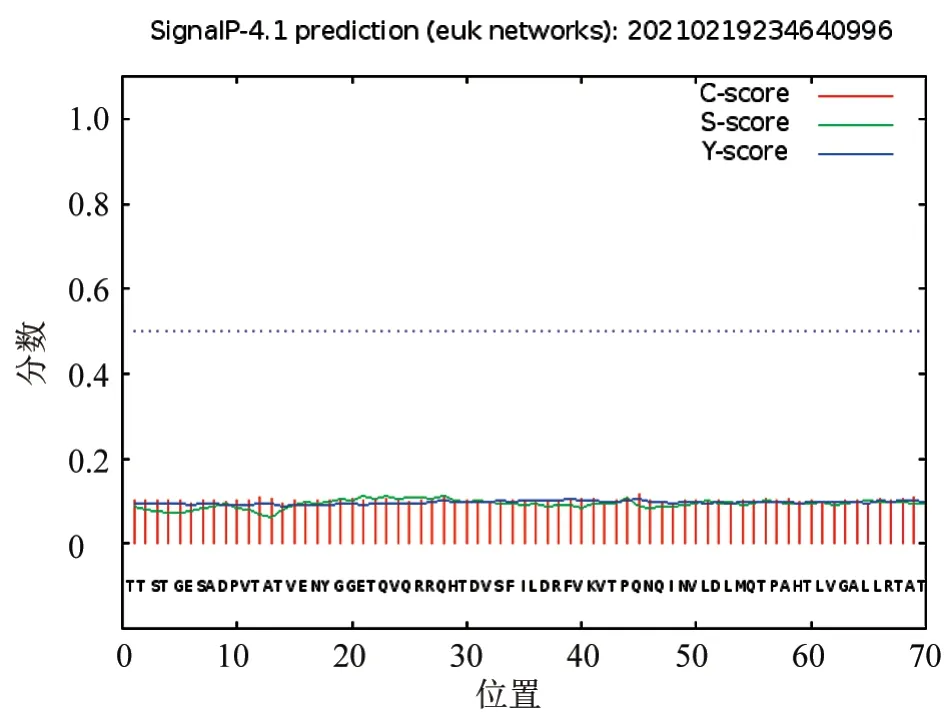
图3 VP1 蛋白信号肽分析结果
2.3 二级结构预测
使用SOPMA 软件预测VP1 蛋白的二级结构,发现α-螺旋在VP1 蛋白二级结构中占比为28.17%,β-折叠占比为23.00%,而占比最多的区域为无规则卷曲(41.78%),占比最少的为β-转角(7.04%),具体见图4。该型与不同拓扑型O/CHA/2/99 相比,α-螺旋占比高0.94%,β-折叠占比高4.22%,O/CHA/2/99 占比最多和占比最少的区域也为无规则卷曲和β-转角。
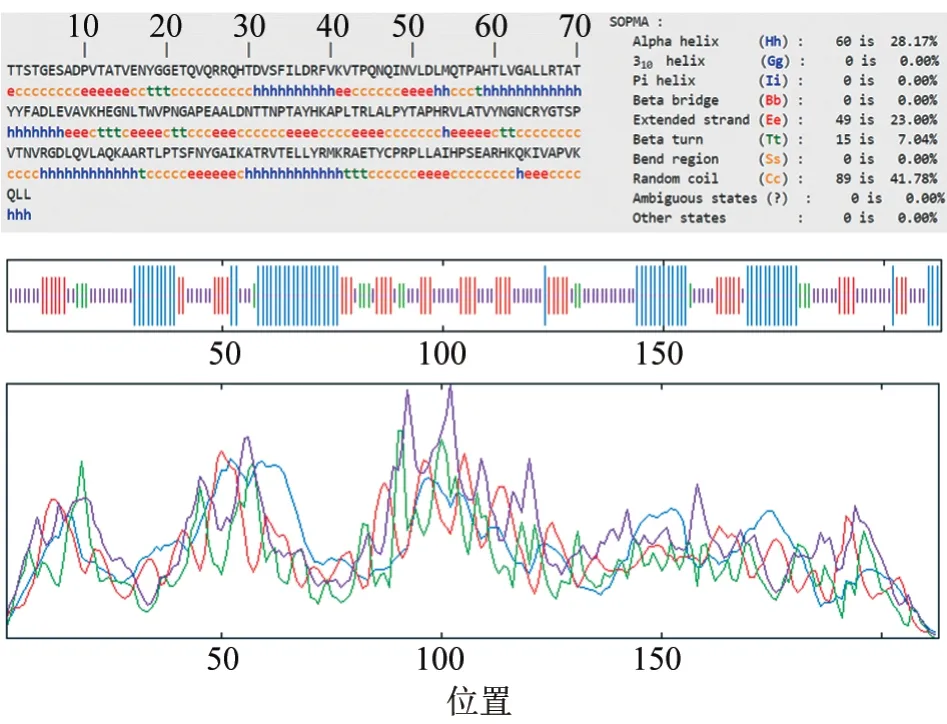
图4 SOPMA 软件预测VP1 蛋白二级结构结果
采用DNAstar Protean 软件中的Gamier-Robson 和Chou-Fasman 两种方法,对VP1 蛋白的二级结构进行预测。两种方法预测的VP1 蛋白结构α-螺旋、β-折叠、转角和无规则卷曲区域分布如表1 所示。

表1 DNAstar Protean 软件预测VP1 蛋白二级结构结果
综合上述两种软件对VP1 蛋白二级结构的预测分析结果,SOPMA 软件预测VP1 蛋白的二级结构中无规则卷曲占比最高,占比最少的区域为β-转角;DNAstar Protean 软件预测的二级结构中β-折叠占比最高,无规则卷曲区域和转角区域占相对较少。α-螺旋和β-折叠被认为是蛋白的骨架区域,由于两种结构的特殊性,这两个区域不利于形成位点。在VP1 蛋白的二级结构预测结果中,虽然β-折叠占比较高,但是结合其亲疏水性,预测VP1蛋白上存在多个抗原位点。
2.4 三级结构预测
使用SWISS MODEL 软件同源构建VP1 蛋白三级结构模型(图5)。选择5nem.1.A 为模板,发现序列相似度为95.24%,覆盖值为0.99,GMQE(全球模型质量估计)值为0.85,QMEAN值为-3.66。GMQE 是一种基于目标模板对准结合性质的质量估计,所得的分数以0~1 的数字表示,反映了该对齐方式和模板构建模型的预期准确性以及目标的覆盖范围,越接近1 表示模型越接近试验结果。QMEAN 对模型的质量估计是基于蛋白模型的局部和全局计分,分值范围为-0.4~0,越接近0表明模型结构与相似大小的试验结构之间具有良好的一致性。Phyre2 软件预测VP1 蛋白三级结构,同源模板为d1qqp1,188 个残基(88%的序列)通过单个评分最高的模板,以100%的置信度建模。结果(图6)显示,VP1 蛋白二级结构以β-折叠(占比43%)为主,其次是α-螺旋(占比12%),与DNAstar 软件预测的二级结构结果一致。phyre 同源建模是蛋白质结构数据库中寻找未知蛋白结构的同源伙伴,将同源蛋白质的结构优化,构建出预测蛋白质的三维结构,选择1 个单体蛋白来建模[16]。
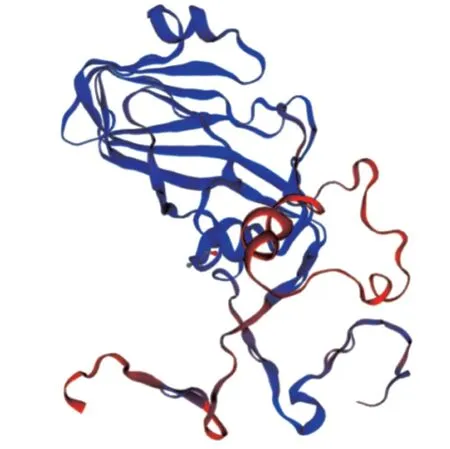
图5 SIWSS MODEL 预测的VP1 蛋白三级结构模型
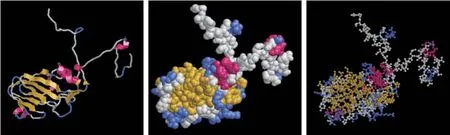
图6 Pryre 预测的VP1 蛋白三级结构模型
2.5 B 细胞表面抗原表位预测
ABCpred 软件是通过基于神经网络的一种预测方法[17]。通过ABCpred 软件预测VP1 蛋白B 细胞表位,序列得分越高,表明作为表位的概率越高。选择得分最高的前10 条序列,结果如表2 所示。
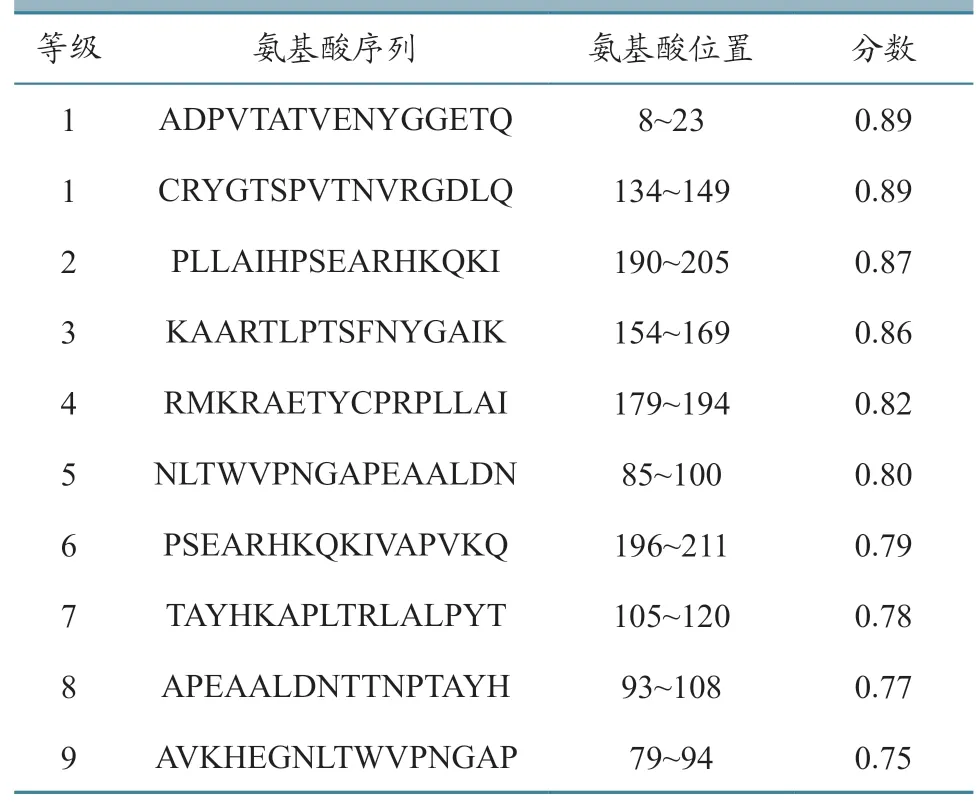
表2 ABCpred 预测VP1 蛋白B 细胞抗原表位结果
IEDB 在线软件通过结合6 个方面的预测确定VP1 蛋白的抗原表位,分别为线性表位、β-转角、表面可及性、骨架区柔韧性、抗原指数和亲水性等。预测结果如图7 所示。结合ABCpred 软件分析结果,排除α-螺旋、β-折叠、疏水区等不易形成表位的区域综合分析预测,确定了3 个B 细胞表位,分别是8~23 位(ADPVTATVENYGGETQ)、135~149 位(RYGTSPVTNVRGDLQ)和193~205位(AIHPSEARHKQKI)。
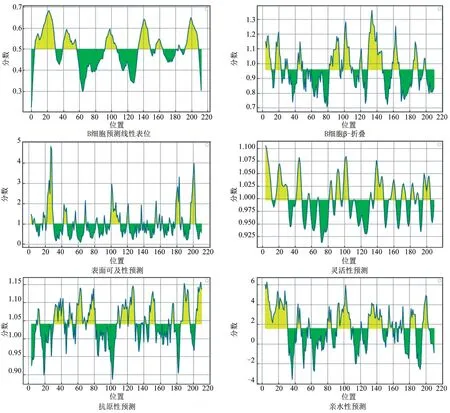
图7 IEDB 软件软件预测VP1 蛋白B 细胞表位抗原结果
2.6 T 细胞表面抗原表位预测
抗原表位是抗原分子中诱导机体产生特异性免疫应答的特殊化学基团,主要分为B 细胞抗原表位和T 细胞抗原表位,而T 细胞抗原表位又分为细胞毒性T 细胞(cytotoxic tlymphocyte,CTL)表位及辅助性T 细胞(helper T cell,Th)表位[18]。本研究综合氨基酸残基亲水性、抗原性、可及性等预测抗原表位,随后采用SYFPEITHI 软件和IEDB等在线工具排除α-螺旋和β-折叠不易形成表位的结构,与位于无规则卷曲处的氨基酸序列对比筛选预测出了T 细胞、B 细胞抗原表位,通过对比后得到发现CTL 细胞表位和Th 细胞表位是VP1 蛋白两个主要的T 细胞表位,预测VP1 有10 个CTL细胞抗原表位,10 个Th 细胞抗原表位,最后筛选出了VP1 蛋白5 个CTL 优势表位和5 个Th 优势表位(表3~4)。
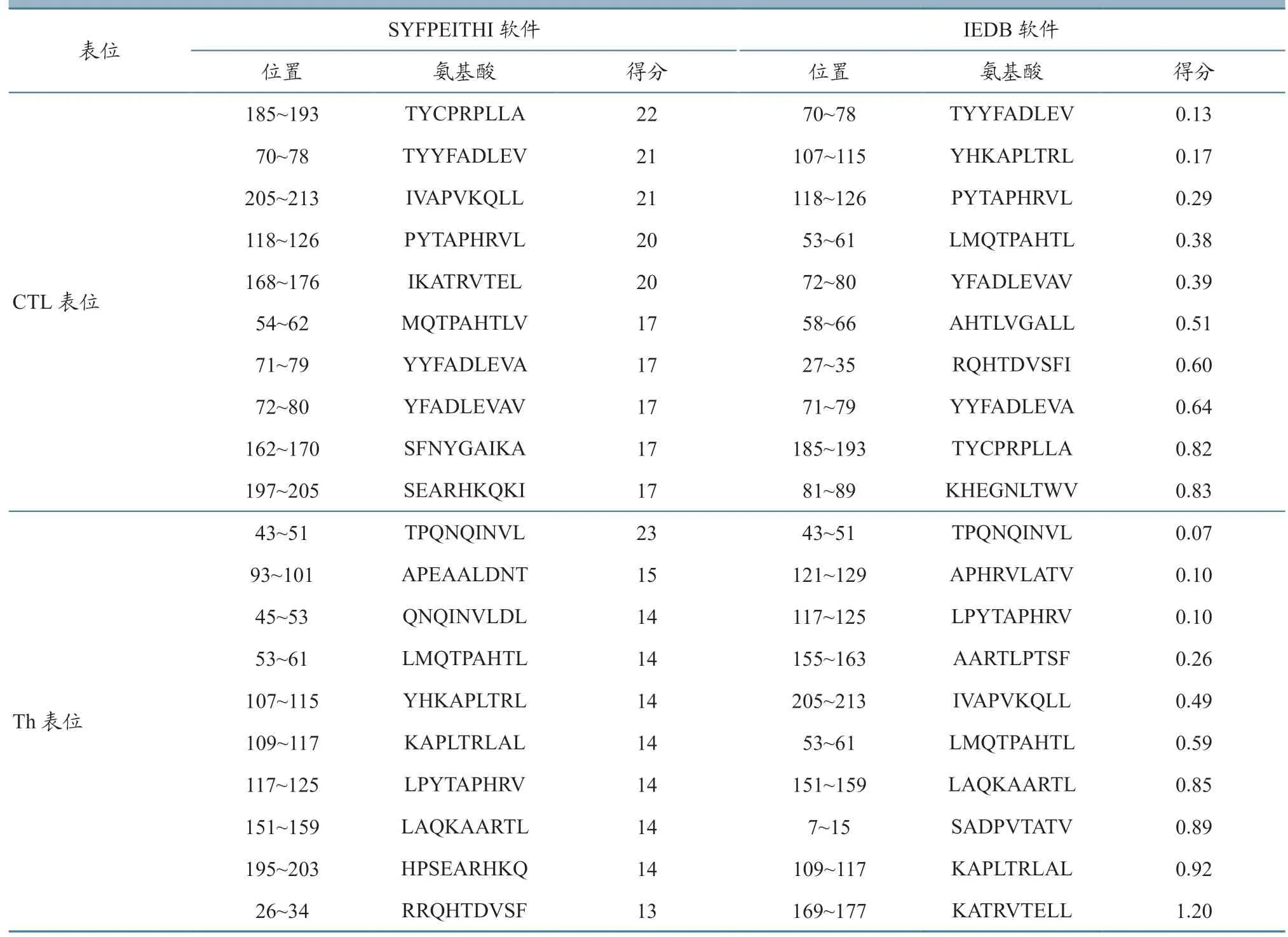
表3 T 细胞表面抗原表位预测结果
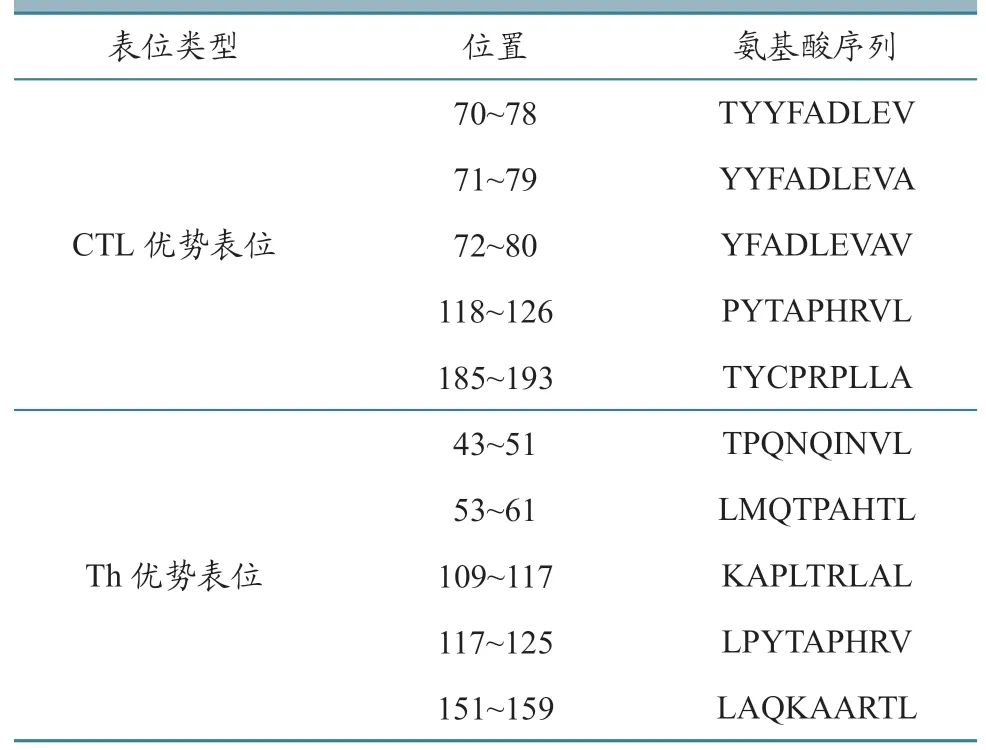
表4 T 细胞优势抗原表位预测结果
3 讨论
FMD 是偶蹄动物的一种高度传染性疾病,猪、牛、羊以及许多野生物种对其均易感。FMD 影响世界许多地区,经常导致大范围流行[19]。在以农业为主要支柱产业的国家,FMD 的暴发严重影响粮食安全和发展。FMDV 分为7 个血清型,目前国内主要流行的是O 型和A 型。我国周边FMD疫情不断,导致多种毒株由境外传入,比如O/MESA/PanAsia、O/SEA/Mya-98 和A/ASIA/Sea-97 毒株等。从我国周边流行的病毒类型、流行频率和循环区域位置判断,O/PanAsia-2、A/Iran-05、Asia1/Sindh-08 和 O/Ind-2001 等毒株未来对我国养殖业威胁依然较大,因此制备亚单位疫苗将有利于我国FMD 的防控[1]。
本研究通过生物信息学软件,分析O/PanAsia株VP1 蛋白的理化特性,发现VP1 蛋白总体上是一个结构稳定的亲水性蛋白,不存在跨膜结构域且该蛋白无信号肽存在。该蛋白的二级结构主要由β-转角构成,其次为β-折叠。通过SWISS MODEL 软件构建三级结构,发现VP1 蛋白具有多个可以形成表位的空间结构。
表位预测在公共卫生和基础科学研究中具有重大意义。它适用于与表位相关的研究,比如亚单位疫苗候选多肽研究、自身免疫性疾病研究、过敏治疗、蛋白质结构研究和实验设计等[20]。当前,在疫苗研究中使用生物信息学分析蛋白结构和预测表位成为大多数实验室的首选方案,与常规试验相比,采用生物信息学技术能够使预测结果更加准确和高效。曾江勇[21]预测猪FMDV Hankou/99 株结构蛋白的二级结构及B 细胞抗原表位,表明该病毒除了VP1 区域,VP2 和VP3 也存在抗原表位。杨鑫等[22]通过预测Asia Ⅰ型VPl 结构蛋白的B 细胞抗原表位,然后与国内报道序列的二级结构进行分析和比较,发现该毒株VP1 蛋白的抗原表位区域具有一定的差异。高瞻等[11]构建了FMDV O/Ind2001 株VP1 蛋白的二级结构分子模型,并结合生物信息学技术预测了基于H2-Ld 及H2-Kd 两种不同 MHC 限制性T 细胞表位及B 细胞抗原表位。与O/Ind2001 株比对VP1 蛋白的抗原表位发现,虽然部分抗原表位有一定的同一性,但不同拓扑型毒株之间的VP1 蛋白结构与表位还是存在差异。VP1基因在进化或传播过程中容易发生突变,这可能是FMD 难防难控的原因之一。因此,需要分析FMDV 不同拓扑型的优势抗原表位,进行多方位预测。鉴于FMDV 不同毒株之间不存在交叉免疫性,因此需要针对本地区流行毒株制备疫苗。
有研究[13]表明,α-螺旋和β-折叠区域位于蛋白内部,易形成稳定结构,较难形成表位。而β-转角和无规则卷曲区域较为松散,是很好的形成抗原表位区域。亲水性强以及可及性和可塑性较好的区域也是形成表位的优势区域,因此VP1 蛋白的结构显示出其具有很好的表位预测价值。
ABCpred 软件和IEDB 软件都具有很较高的预测精度,综合两者分析结果,预测VP1 蛋白的B 细胞表位为8~23 位、135~149 位和193~205 位氨基酸。本研究使用SYFPEITHI 和 IEDB 软件分析了VP1 蛋白的T 细胞抗原表位,发现CTL 细胞表位和Th 细胞表位是VP1 蛋白两个主要的T 细胞表位,预测VP1 有10 个CTL 细胞抗原表位,10个Th 细胞抗原表位,表明VP1 蛋白具有较好的免疫原性,最后筛选出了VP1 蛋白5 个CTL 优势表位和5 个Th 优势表位。而A 型病毒株AF72 表位VP1a 和VP1d 为病毒株AF72 结构蛋白VP1 的优势B 细胞表位,21~40 位氨基酸是T 细胞表位,引发细胞免疫应答;141~160 位及200~213 位氨基酸为B 细胞表位,引发体液免疫反应[15]。A 型和O 型为FMDV 的不同亚型,因此其优势细胞表位也不同。通过生物信息学工具对VP1 蛋白的B 细胞和T 细胞抗原表位进行预测,证实VP1 蛋白能够引起较强的CTL 表位免疫反应。该研究尽管仍需试验进行验证,但为预测抗原表位,选择合适肽段,继而制备多肽疫苗提供了理论依据。
猜你喜欢
科学大观园(2022年2期)2022-01-23
温州医科大学学报(2019年4期)2019-04-28
中国免疫学杂志(2017年1期)2017-01-17
动物医学进展(2015年10期)2015-12-07
西南医科大学学报(2015年1期)2015-08-22
畜牧兽医学报(2015年3期)2015-07-05
医学研究杂志(2015年6期)2015-07-01
癌变·畸变·突变(2015年3期)2015-02-27
特产研究(2014年4期)2014-04-10
郑州大学学报(理学版)(2014年3期)2014-03-01
