
氧化石墨烯环境下C-S-H生成分子动力学研究
2021-11-08 03:19潘文浩何淼弘
沈阳建筑大学学报(自然科学版) 2021年5期
郭 凯,潘文浩,何淼弘,刘 琳
(1.沈阳建筑大学土木工程学院,辽宁 沈阳 110168;2.沈阳建筑大学材料科学与工程学院,辽宁 沈阳 110168)
水化硅酸钙(C-S-H)是水泥水化的主要产物,占据的体积分数在60%以上,是水泥基材料中力学性能和耐久性能的主要来源[1]。近些年来纳米技术的发展为水泥基材料的改性提供了一种新的思路。氧化石墨烯(GO)具有巨大的比表面积、优异的力学性能、水溶性能等特点,含有羟基(-OH)、环氧基(-O-)等众多活性官能团,是一种热门的水泥基改性材料[2-3]。GO的掺入可以促进水泥水化,增加水泥水化产物密实度,从而提升水泥基材料的抗拉、抗折性能及耐久性能[4-6]。但由于仪器设备和技术手段的限制,纳米材料在纳观尺度下对水化晶体形貌特征的影响和离子作用是无法用试验的手段准确表征的。
分子动力学模拟是一种基于力场的数值计算方法,可以实现对水化分子的结构和动力学特性在原子、分子尺度上的预测,是非常理想的材料微观模拟的手段之一。利用这种方法,H.Manzano等[7]提出了水分子的分离以及大量羟基官能团的形成机理,较为准确地表征了水合离子在C-S-H凝胶纳米孔洞中的结构、能量与动力学特性[8],建立了不易被描述的C-S-H模型,解析了无机C-S-H凝胶/有机聚合物复合体系的微结构特性与性能提升机理[9]。笔者采用反应分子动力学的方法,参照氧化硅凝胶过程模拟研究早期C-S-H的生成过程,通过在反应体系两侧加入一定宽度狭缝的GO片层,模拟纳米尺度的限制空间环境下GO对C-S-H生成过程与结构变化的影响。
1 研究方法
1.1 C-S-H结构分析
在现阶段C-S-H的原子结构并未被完全认识,也没有统一的模型。氧化硅玻璃制备中溶胶凝胶的过程,主要的化学反应是硅之间羟基的聚合,这种硅链结构的聚合反应本质上与C-S-H凝胶生成过程中水泥离子溶解以及离子之间键合生成沉淀的过程是极为相似的。目前参照氧化硅凝胶过程的模拟,采用反应分子动力学来研究C-S-H早期生成过程的方法,已经被成功地用于研究硅酸盐材料和水之间的反应活性[10-11]。通过计算每个原子的偏配位数,用所有原子对的偏径向分布函数,把第一个峰后的局部最小值作为截断半径定义为原子对的第一配位壳层。统计每个原子在第一配位壳层范围内近邻原子的个数就可以得到与其对应原子种类的偏配位数。采用这种统计方法,可以计算得到结构中的桥位氧原子(Bridging oxygen atoms,BO),即与两个硅原子相连的氧原子;非桥位氧原子(NBO),即只与一个硅原子相连的氧原子。通过统计识别出的BO和NBO,可以进一步定义不同种类的硅原子(Qn,n=0~4),Qn即为与n个BO相连的硅原子。如Q1表示与硅原子相连的4个氧原子中有1个氧原子为BO,另外3个氧原子为NBO。在凝胶反应之前所有硅原子均为Q0,聚合反应开始后两个Q0单体聚合为Q1二聚体,即随着Q0的消耗,Q1、Q2以及少量Q3和Q4摩尔分数开始增加,所以可以通过桥位氧原子与硅原子的比值n(BO)∶n(Si)来描述氧化硅硅链结构胶体中总体的聚合度[12-14],并且可以通过n(BO)∶n(Si)随时间变化的曲线得到聚合的反应速率。
1.2 模型构建与模拟方法
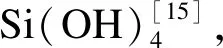
采用MaterialsStudio软件建立初始模型,首先将Si(OH)4分子按周期性排布在立方体盒子中,并随机投放Ca(OH)2和H2O,各方向均采用周期性边界条件,并对模型进行初步结构优化。使用Lammps软件对模型进行研究。力场采用ReaxFF-Fogarty力场,粒子的运动采用速度Verlet算法求解,时间步长设定为0.25 fs。使用正则系综(NVT)对模型进行动力学模拟,使初始体系在300 K温度下驰豫100 ps,阻尼系数确定为25 fs。随后再在等温等压系综(NPT)下300 K温度、压强为0条件下驰豫500 ps,阻尼系数为250 fs。为加速计算,通过提高反应温度的方式加快反应过程[15,17-18]。模型反应温度设定为2 000 K,在NVT系综下恒温进行,设定模拟步数200万步,总时长500 ps。最后,对生成凝胶的系统逐步降温到300 K,并在压强为0的NPT系综条件下驰豫100 ps,得到C-S-H,结束模拟。
石墨烯初始模型来源于系统自带的石墨晶体,通过在x、y方向上复制得到a=3.936 nm,b=3.936 nm的石墨烯片层,根据不同含氧基团对水分和离子吸附特性[19],模拟在石墨烯表面添加羧基(-COOH)形成GO片层,设置片层间距Z为4 mm,氧原子的摩尔分数为50%。在片层中放入与模型一致的分子反应物溶液,采用同样的模拟程序和体系参数,获得早期C-S-H凝胶晶体模型。所有模型构建参数如表1所示,C、O、H、Ca和Si等原子形成的初始体系模型如图1所示。

表1 模型体系组分及相关参数Table 1 Components and related parameters in model
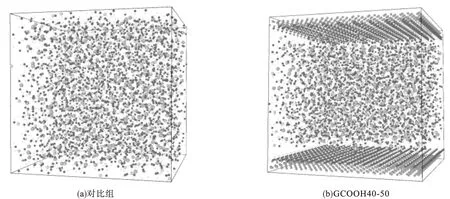
图1 体系初始模型Fig.1 Initial system of model
2 结果与分析
图2为C-S-H生成模型切片。经反应陈化后可以看出,两个体系内均有硅氧链式和环式结构出现,说明生成了早期C-S-H晶体结构。通过详细分析体系内的相关参数,可以对比不同体系的差异。

图2 C-S-H生成模型切片Fig.2 Slices showing the atomic configurations of C-S-H
2.1 C-S-H结构演变
氧、硅原子在模型中的反应变化如图3所示。随着反应时间的增加,n(BO)∶n(Si)的值不断提高,反映了体系整体聚合度在持续提高,在陈化反应后期逐渐趋于稳定。

图3 n(BO)∶n(Si)变化曲线Fig.3 Variation curve of n(BO)∶n(Si)
对比BO变化曲线,无GO的对比组反应开始时BO数量为0,反应过程稳定,较早达到平衡态。加入GO狭缝的模型中,驰豫过程结束后已有少量BO生成,反应开始时n(BO)∶n(Si)约为0.1,这也说明在纳米狭缝的位阻作用下,原子的碰撞概率得到较大提高,体系更易聚合、形成链式分子结构。模拟结束时,对比组n(BO)∶n(Si)的值为0.38,而GCOOH40-50体系内BO数量明显增加,n(BO)∶n(Si)的值最终达到0.63,较对比组提高65.8%。对比两组BO/Si曲线的初始斜率,其对应了陈化反应的聚合速率。在前期,对比组与GCOOH40-50反应速率较为接近,并没有明显差异。反应中期,纳米狭缝的调控作用使聚合反应得以持续进行,形成了更多数量的BO。
统计硅原子键连BO的数量,将硅原子分为Q0~Q4,比较体系中Qn的变化曲线如图4所示,最终Qn的摩尔分数分布如图5所示。可以看到,参照组在反应开始时只含有Q0,后续反应中逐步生成Q1、Q2以及少量Q3,在模拟进行至200ps时体系已经基本稳定趋于收敛,最终Q1、Q2、Q3单元占比分别为46.29%、13.43%、5.56%,二聚体的Q1单元体量最大,并且体系内几乎不存在Q4单元。
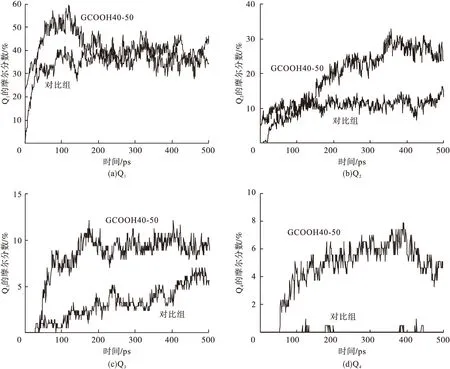
图4 Qn摩尔分数变化曲线Fig.4 The curve of the mole fraction change of Qn

图5 各体系Qn的摩尔分数分布Fig.5 Mole fraction distribution of Qn in each system
在GCOOH40-50体系中,反应开始时已经存在少量Q1,这是在前期驰豫过程中形成的,之后随着反应进行Q1快速增加,同时Q2体量也在稳步提高,Q3的出现则相对较晚。在反应进行至100~150 ps时,Q1占比达到最高值,之后Q2、Q3单元开始大量出现,Q1所占比例随之下降。反应在500ps结束时,各单元占比分别为34.72%、23.61%、8.79%,虽然Q1仍为主相,但相较于对比组Q2、Q3已经有了极大的提高,同时值得注意的是,体系内稳定的存在约为4%的Q4单元。
2.2 径向分布函数分析
径向分布函数(Ranial Distribution Function,RDF)是描述距参考原子一定距离r内的目标粒子密度变化的函数,可以反应模型中任意原子一定范围内特定粒子出现的几率g(r)和排布变化,是分子动力学中常用的一种分析方法。其与X-Ray小角度衍射试验得到的函数互为傅里叶变化,分析函数为
(1)
式中:r为距离中心原子半径;n(r)为球壳范围内原子数;ρ为晶体密度;δ(r)为球壳厚度。
通过计算可以得到Si-O、Si-Si、Ca-O和Ca-Ca 4类原子对的径向分布函数(见图6)。从图6(a)可以看出,所有模型的Si-O键函数曲线均是在0.16 nm处出现峰值点,但峰值强度具有较大差异,相比于对照组,GCOOH40-50模型的峰值有了显著的提高。这说明在4 nm狭缝的GO环境下模型中硅原子和氧原子键连数量增加,密度明显提高。函数曲线在0.4 nm处出现的馒头峰对应的是模型体系中非键连的硅、氧原子,模型的峰值高度差别不大从。图6(b)可以看出,Si-Si的径向分布函数,硅原子与硅原子通过桥位氧原子形成链接,曲线主位峰值点出现在0.32 nm处,GCOOH40-50模型的峰值强度明显高于对照组,这说明在纳米GO的作用下桥位氧原子的增加使硅原子的聚合得到了有效的提高。Si-Si函数曲线在0.5 nm处出现馒头峰,两曲线峰值仍比较接近。从6(c)可以看出,Ca-O的径向分布函数,GCOOH40-50模型的高聚合度使曲线0.24 nm处的峰值点高于对照组,Ca-O键密度有明显提高。图6(d)中GCOOH40-50模型的Ca-Ca分布仍然较高,因为钙原子之间并不存在键合,说明体系内无关于聚合度的情况下,GO的作用仍使溶液中的钙原子出现了明显的聚集。
图6 原子径向分布函数Fig.6 Atomic radial distribution function
2.3 化学组分的分布
对模型中钙原子、硅原子在z轴方向上的分布进行统计,可以得到钙、硅的分布曲线(见图7)。进而分析模型体系中生成C-S-H密度的差异。图7(a)中参照组钙、硅的分布曲线可以看到原子在z轴方向上分布较为均匀,各个位置钙硅物质的量比基本与初始体系中的1.5保持一致,在中部2.5~3.0 nm处出现了原子分布的数量峰值,此区域即C-S-H富集区,这也说明C-S-H的生成的确具有聚集效应,倾向于形成局部团簇。图7(b)GCOOH40-50模型中由于存在约为0.4 nm的GO片层,所以原子分布在0.4~3.7 nm内,钙、硅的分布曲线的峰值出现了明显的向一侧GO片层的偏移,钙原子分布的富集区域出现在0.7~1.2 nm,硅原子富集区域在1.0~1.3 nm,说明GO对钙、硅原子均有吸附作用,且对钙原子的吸附更为明显,在局部形成了更高钙硅物质的量比的区域。通过不同钙硅物质的量比的C-S-H凝胶聚合模拟可知[11],不同钙硅物质的量比条件下C-S-H的生成势垒存在差异,C-S-H生成焓会随钙硅物质的量比的提高而较少,而活化能会随着钙硅物质的量比的增加先下降后提高,在1.8时达最小值,这是由于继续提高钙原子数量将会诱导生成更多的NBO,降低胶体总体的聚合度。所以含有GO的模型中合适的钙原子富集使体系局部钙硅物质的量比增大,在此浓度下钙原子催化了早期凝胶聚合反应[16,19],并且起到了隔离纳米空间的作用,使得Si(OH)4单体富集,提高了碰撞几率、提高了聚合反应程度。
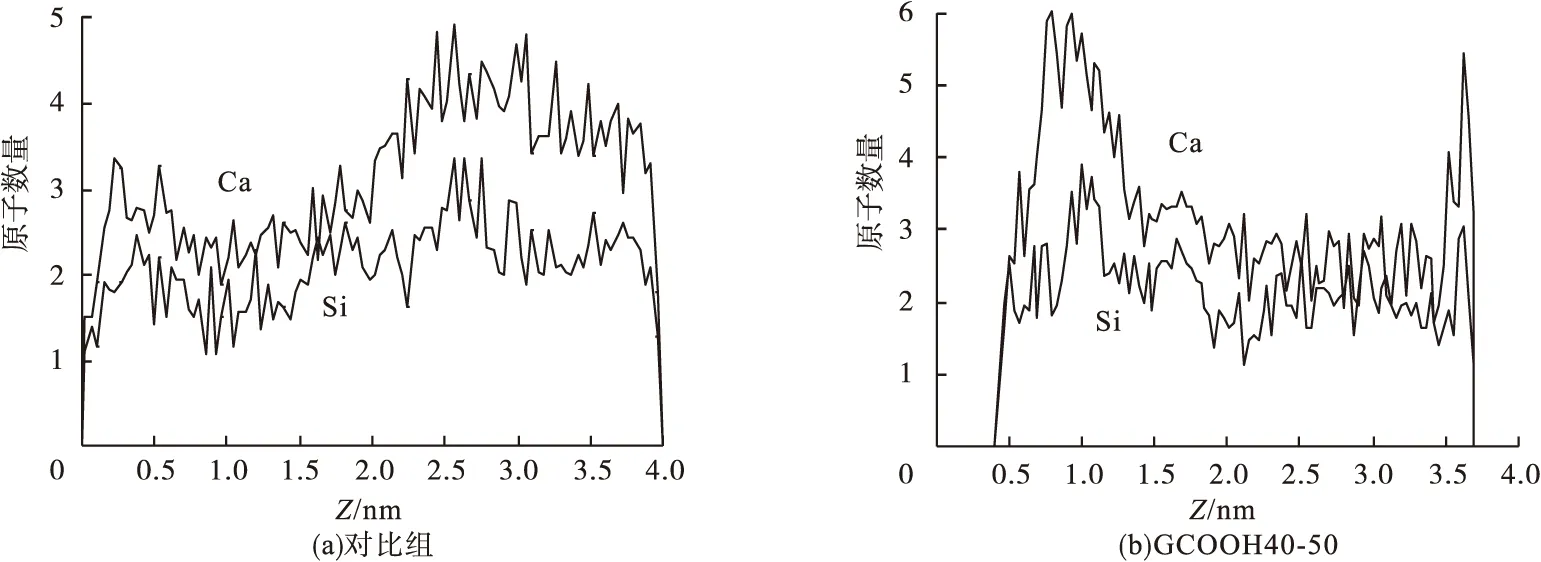
图7 沿z方向Ca和Si原子分布Fig.7 Distribution of Ca and Si atoms in Z direction
2.4 Qn结构的分布
凝胶的原子结构很大程度上决定了C-S-H的化学组分,因此通过探究Qn单元沿z轴方向的分布就可以表征C-S-H在体系中的结构变化。图8为沿z方向Qn单元分布。图8(a)为对照组Qn单元的分布情况,可以看到Q1、Q2单元均在2.5 nm附近出现较多数量聚集,Q3单元数量较少,随机分布,说明C-S-H多为短链结构,汇聚位置明确,体系中还存在较多Q0单元,说明系统中含有一定量C-H,体现出早期C-S-H体系聚合度较低,分布不均匀。图8(b)展示了GCOOH40-50模型中Qn单元分布,Q0单元摩尔分数较参照组更低,Q1单元整体分布相对均匀,在1.0 nm处数量较多但与其他位置相差不大,Q2、Q3单元在此位置数量较为聚集,这与硅原子分布图7(b)中的富硅区域是一致的,说明高链接的C-S-H结构易出现在高钙硅物质的量比的富硅区域,体系中有Q4单元出现在各区域,在1.0 nm处摩尔分数最大,也证实了这一点。
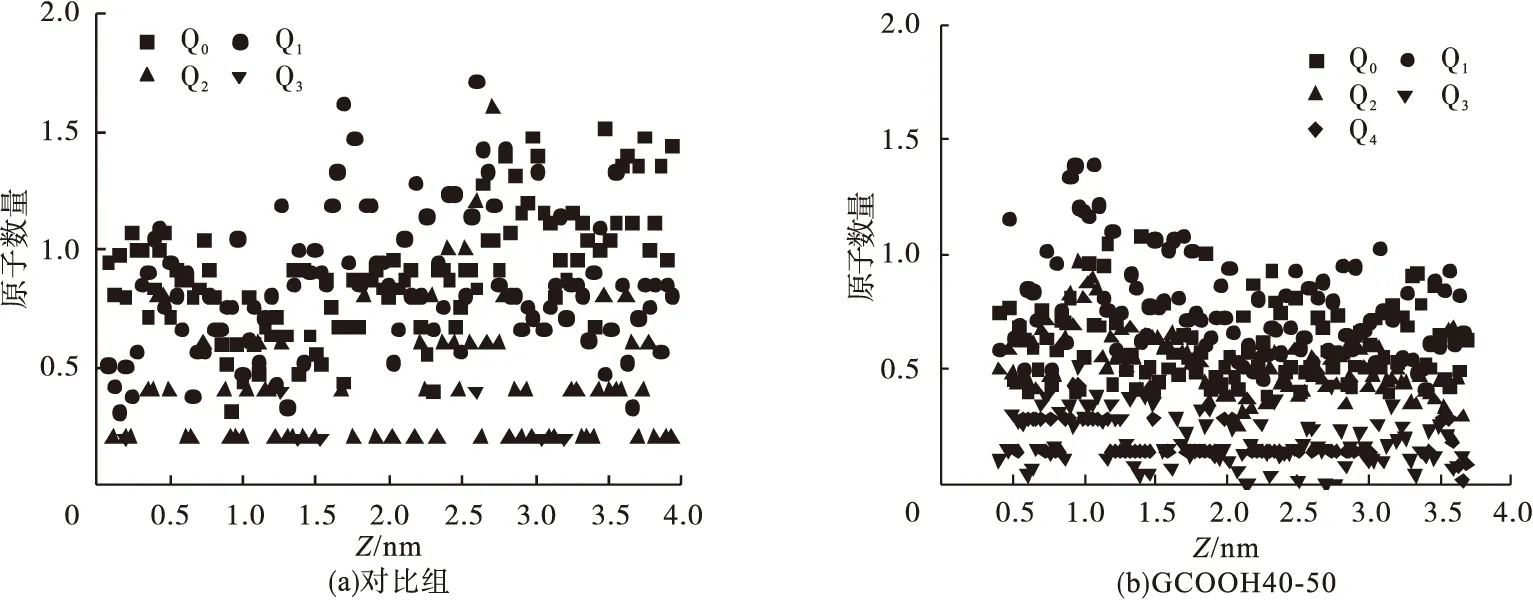
图8 沿z方向Qn单元分布Fig.8 Qn element distribution in Z direction
对比两组模型,GCOOH40-50中GO在一定程度提高了聚合单元数量,但对结构最大的改变在于对于聚合物位相的偏移以及聚合物聚合度的提高。
3 结 论
(1)反应分子动力学的方法能够较为准确地反映早期C-S-H的生成过程,将体系置于纳米GO环境下也得到了完整的早期C-S-H结构。
(2)在纳米GO环境作用下,陈化反应前期GO对体系的反应速率并没有明显的影响。但GO的限域作用,使原子的碰撞几率提高,反应在中后期持续进行,高链接性Qn单元也随之产生。
(3)GO中的含氧基团可以吸附钙、硅原子,使体系内局部形成钙、硅原子富集区,降低了聚合势垒,使C-S-H聚合度得到了提高。
猜你喜欢
中国造纸(2022年3期)2022-07-21
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
椰城(2021年12期)2021-12-10
食品与发酵工业(2021年14期)2021-08-02
少儿科学周刊·少年版(2021年22期)2021-01-17
物理学报(2017年21期)2017-11-10
腐蚀与防护(2016年7期)2016-09-14
天然产物研究与开发(2014年1期)2014-04-27
