
Radio-HPLC分析方法测定治疗用碘[131I]化钠胶囊的放射化学纯度
2021-11-08 12:22程缤雁李洪玉
核化学与放射化学 2021年5期
杨 柳,程缤雁,付 博,姜 华,李洪玉,张 云
原子高科股份有限公司,北京 102413
放射性药品(radiopharmaceuticals)是指含有一种或几种放射性核素供医学诊断和治疗的药品[1]。放射化学纯度(radiochemistry purity,简称放化纯度)是指某一特定化学形式的放射性核素的放射性量占该核素总放射性量的比例(Y)。放化纯度作为放射性药品的关键质量控制项目,在工艺研究阶段的配方优化和注册上市后的质量控制中均起着非常重要的作用。
治疗用碘[131I]化钠胶囊是可用于甲状腺疾病治疗的胶囊剂型放射性药品,其放化纯度测定方法主要有纸色谱(paper chromatography, PC)法和放射性-高效液相色谱(radio-high performance liquid chromatography, Radio-HPLC)法。PC法是以纸为载体,以纸上所含水分或其他物质为固定相,用适宜的展开剂对放射性样品溶液进行展开的分配色谱法,用合适的放射性检测器测量展开后的色层纸以确定各组分的放射性分布[2]。《美国药典》40版(USP40版)收录的碘[131I]化钠胶囊放化纯度测定采用了PC法[3],明确以尺寸为25 mm×300 mm的色层纸作为固定相、75%(体积比)甲醇作为流动相的展开条件对放射性主峰与杂质峰进行分离,但色层纸展开长度过长影响了检测效率。Radio-HPLC法是在高效液相色谱系统中检测器模块处,将放射性检测器串联于常规检测器(如紫外分光光度计、二极管阵列检测器、电化学检测器等)之后,色谱系统前端色谱柱与流动相相互作用实现各种组分的分离,并流经放射性检测器进行放射性活度的测定以确定各组分的放射性分布[4-5]。Radio-HPLC法中常规检测器可以实现“冷化合物”色谱峰的检测并与各放射性化学成分进行比对从而达到鉴别的目的。《欧洲药典》9.0版(EP9.0版)中治疗用碘[131I]化钠胶囊的放化纯度测定便采用了Radio-HPLC法[6],一方面,该方法具有灵敏度好、准确度高等优点,有利于治疗用碘[131I]化钠胶囊放化纯度分析过程中放射化学杂质的准确测定;另一方面,该方法将紫外分光光度计与放射性检测器有机结合,从而实现了治疗用碘[131I]化钠胶囊放化纯度分析过程中放射化学杂质的定性鉴别。
2021年4月,原子高科股份有限公司研制的治疗用碘[131I]化钠胶囊获得国家药品监督管理局批准上市。该胶囊制剂的内容物包含一定放射性活度的碘[131I]、抗氧剂A和填充剂B,抗氧剂和填充剂的种类与EP9.0版收录的治疗用碘[131I]化钠胶囊不同;另外,与口服溶液剂型和注射剂型相比,胶囊剂型的放射性药品放化纯度方法学研究需考虑的影响因素较多。因此,本工作拟从胶囊的前处理方法、辅料对分析方法的影响以及放射化学杂质的研究等方面入手,建立测定治疗用碘[131I]化钠胶囊放化纯度的Radio-HPLC分析方法,为建立合理的放射性药品质量标准提供技术参考。
1 实验部分
1.1 试剂和仪器
氯化钠(分析纯)、碳酸氢钠(分析纯)、甲醇(分析纯)、磷酸(优级纯)、乙腈(色谱纯),国药集团化学试剂有限公司;正辛胺(分析纯)、碘化钾标准物质、碘酸钾标准物质,阿拉丁试剂有限公司;灭菌注射用水,石家庄四药有限公司;碘[131I]化钠口服溶液,原子高科股份有限公司;十八烷基硅烷键合硅胶填充的色谱柱(4.6 mm×250 mm×5 μm),美国Agilent公司;0.45 μm滤膜,北京化大膜材料厂;砂芯抽滤装置,颇尔公司。
1260高效液相色谱仪(包括四元泵、紫外检测器、柱温箱和自动进样器),美国Agilent公司;LB514放射性流量检测仪,德国伯托公司;AB135十万分之一电子分析天平,梅特勒-托利多公司;超声波清洗仪,上海鲁硕实业有限公司;YP502N千分之一电子天平,上海菁海仪器有限公司;CRC-55tR活度计,美国CAPINTEC公司。
1.2 色谱条件
采用十八烷基硅烷键合硅胶为填料的色谱柱(4.6 mm×250 mm×5 μm,pH适用范围为2~9);以5.85 g/L氯化钠溶液(加入0.65 mL正辛胺并用稀磷酸调pH至7.0)-乙腈(V∶V=20∶1)为流动相,经0.45 μm滤膜滤过后超声去除气泡后使用;放射性流量检测仪(BGO检测池,15 μL)与紫外检测器(检测波长为220 nm)串联,柱温为25 ℃,进样量为20 μL,运行时间为40 min,以1.5 mL/min流速进行等度洗脱。以下若无特别说明,均按照此色谱条件进行实验。
1.3 溶液的制备
1.3.1对照溶液 分别取碘化钾26.22 mg和碘酸钾24.50 mg,分别置于1 L量瓶中,加水溶解并稀释至刻度,摇匀,得到碘化钾和碘酸钾的储备液;再精密量取碘化钾和碘酸钾的储备液各1 mL,分别置于10 mL量瓶中,加水稀释至刻度,即得含碘化物2.0 mg/L的对照溶液1和含碘酸根2.0 mg/L的碘酸钾溶液,二者等体积混合得到对照溶液2。
1.3.2载体溶液 取碘化钾1 g、碘酸钾2 g与碳酸氢钠10 g,加1 000 mL水溶解制成。
1.3.3样品溶液 取1粒治疗用碘[131I]化钠胶囊,将内容物溶解于10 mL水中,超声助溶,静置后,取上层清液,用载体溶液稀释一倍,作为样品溶液。
1.3.4阴性对照溶液 按照治疗用碘[131I]化钠胶囊的处方工艺组成,取抗氧剂A、填充剂B等制剂辅料按处方比例制备不含碘[131I]化钠溶液的胶囊,按1.3.3节方法制备阴性对照溶液。
1.4 判定标准
在样品溶液的放射性色谱图中,应满足以下两点:(1) 碘[131I]化钠的放化纯度按放射性峰面积归一化法计算应不得低于95%;(2) 放射性主峰的保留时间与对照溶液1中碘化物峰的保留时间相差不超过10%。
2 结果和讨论
2.1 辅料对分析方法运行时间的影响
以流动相作为空白参考,进样色谱系统进行分析,考察对照溶液1、抗氧剂A溶液和填充剂B溶液的主峰保留时间,以确定适宜的方法运行时间。碘化物的色谱峰近似于对称形正态分布,填充剂B在该条件下无紫外吸收,而抗氧剂A的色谱峰拖尾严重,运行时间为40 min才能保证出峰完全以至于不会影响下一针的样品分析;对比主峰保留时间,抗氧剂A和填充剂B在碘化物色谱峰位置处无干扰,即不会影响放射性主峰碘[131I]化物的鉴别。
2.2 样品溶液制备方法的优化
在治疗用碘[131I]化钠胶囊的制备工艺过程中,碘[131I]药液滴加步骤使胶囊中的填充剂结块,块状物的形成导致内容物溶解效率较低,严重影响了方法的回收率。通过模拟实验发现,在进行样品放化纯度测定前处理过程中,若对胶囊内容物只是简单的加水溶解,不进行任何辅助操作,回收率仅能达到25%左右;而采用超声助溶后回收率可提高至70%以上。
2.3 样品氧化降解实验条件的优化
在研究分析方法的专属性时,需要考察分析方法是否能够有效地将主峰与潜在的杂质峰进行分离。如果治疗用碘[131I]化钠胶囊中抗氧剂量不足,可能会发生因外部氧化作用而引起碘[131I]的降解。该实验采用常见的强氧化剂包括浓硝酸、浓硫酸、双氧水、次氯酸钠、高氯酸、氯酸钾等对治疗用碘[131I]化钠胶囊进行了强降解实验。以碘离子和碘[131I]离子为反应物,分别与一定浓度的浓硝酸、次氯酸钠、高氯酸和双氧水进行反应,待反应完毕经稀释制成测试溶液进样分析,放射性流量检测仪未检测到信号,推测可能是碘离子和碘[131I]离子只被氧化到单质碘(单质碘[131I]),而单质碘(单质碘[131I])在反应过程中已挥发,所以无法检测到。当使用氯酸钾-浓硫酸作为氧化剂体系时:反应之初,溶液体系变为红棕色,随着反应的进行,出现大量的黑色固体,反应至一定时间后,红棕色以及黑色固体消失,反应溶液变得澄清,经稀释制成测试溶液进样分析,可观察到原碘[131I]化物处放射性峰消失,同时产生与碘酸根保留时间一致的放射性峰。通过该降解实验得到的放射性峰为碘[131I]酸根。
2.4 方法验证实验
2.4.1系统适用性 吸取流动相、水、对照溶液1、对照溶液2和阴性对照溶液各20 μL,进行色谱分析。图谱中5.2 min左右的紫外色谱峰为流动相和水色谱图中固有峰位,通过对照溶液1和溶液2的紫外色谱峰情况确定了碘化物和碘酸根的紫外色谱峰位置,阴性对照溶液在碘化物、碘酸根紫外色谱峰处均无干扰,因此对碘[131I]化物、碘[131I]酸根放射性峰的鉴别无影响,对照溶液2中碘化物和碘酸根紫外色谱峰的分离度大于1.5。
2.4.2专属性 对碘[131I]化物溶液进行氧化降解制备得到碘[131I]酸根溶液,对比碘[131I]化物主峰与潜在的降解杂质碘[131I]酸根峰的保留时间,并计算二者的分离度,考察分析方法对治疗用碘[131I]化钠胶囊的放化纯度准确、可靠测定的能力。测试溶液的制备方法如下:
(1) 未破坏样品分析:取1粒治疗用碘[131I]化钠胶囊内容物于10 mL西林瓶中,超声助溶,静置后,取上层清液,作为碘[131I]化物溶液;用载体溶液稀释一倍,作为测试溶液1;
(2) 氧化降解实验样品分析:取一定浓度碘化钾溶液于10 mL反应瓶中,滴入适量碘[131I]化物溶液,再依次加入与碘化钾溶液等体积的9 mol/L浓硫酸以及氯酸钾溶液,密封,温水浴反应一定时间,待反应体系变澄清,即表示碘[131I]化物的氧化降解反应完成。随即取0.2 mL反应液,测得其活度浓度为47.36 GBq/L,加入0.8 mL灭菌注射用水稀释淬灭反应,得到活度浓度为9.62 GBq/L的溶液,取适量再与等体积载体溶液混合均匀,作为测试溶液2;并将测试溶液1与2按一定比例混合,得到同时含碘[131I]化物和氧化降解产物的测试溶液3。
对测试溶液1、2、3进行色谱分析,结果示于图1。从测试溶液1和2的放射性图谱可知,碘[131I]化物和氧化降解产物的放射性峰保留时间分别为7.779 min和2.815 min。从测试溶液3的放射性图谱可知,降解产物放射性峰与主峰碘[131I]化物能够实现完全分离,分离度为31.58,远远满足分离度不小于1.5的要求[7]。根据反应可能的原理推测降解产物为碘[131I]酸根离子[8-9],与对照溶液1和2对比,碘化物和碘酸根紫外峰分别与碘[131I]化物和碘[131I]酸根放射性峰一一对应。以上实验表明,在氧化条件下产生的降解产物碘[131I]酸根对放射性主成分碘[131I]化物测定无干扰,说明所建立的HPLC法测定治疗用碘[131I]化钠胶囊的放化纯度专属性良好。

上图是放射性图谱,下图是紫外图谱
2.4.3线性及浓度测量范围 以一定浓度的碘[131I]化物溶液和碘[131I]酸根溶液作为线性储备液进行稀释,分别得到一系列不同放射性浓度水平的溶液,考察主峰峰面积与放射性浓度成比例关系的能力,并确定分析方法的测量范围。
(1) 碘[131I]化物
线性母液:取一批次碘[131I]化钠口服溶液配制得到活度浓度为1 502.20 GBq/L的线性母液200.0 μL。线性储备液:量取线性母液150.0 μL于色谱用进样瓶内,加灭菌注射用水稀释至0.6 mL,得碘[131I]化物活度浓度约为384.80 GBq/L的线性储备液。线性溶液:分别量取线性储备液5.0、8.0、40.0、100.0、100.0、100.0 μL于色谱用进样瓶中,经稀释后得到一系列碘[131I]化物溶液,其活度浓度为0.37、1.85、9.25、46.25、92.50、185.00 GBq/L,该系列溶液分别与等体积载体溶液混合,即得到碘[131I]化物活度浓度为0.19、0.92、4.62、23.12、46.25、92.50 GBq/L的线性溶液。对这些样品进行色谱分析,每个溶液重复测定2次,记录每个线性点的峰面积,计算其平均值,以碘[131I]化物活度浓度对峰面积进行线性回归,线性曲线示于图2。由图2可知:碘[131I]化物活度浓度在0.19~92.50 GBq/L范围内具有良好的线性关系,线性回归方程为y=161.90x+466.32,线性相关系数r2=0.985。

图2 碘[131I]化物的线性曲线
(2) 碘[131I]酸根
线性母液:氧化降解实验制备得到47.36 GBq/L碘[131I]酸根溶液。线性储备液:量取线性母液200.0 μL于色谱用进样瓶内,加灭菌注射用水稀释至1.0 mL,得碘[131I]酸根活度浓度为9.62 GBq/L的线性储备液。线性溶液:由线性储备液逐级稀释1倍分别得到一系列溶液,溶液活度浓度分别为9.62、4.81、2.40、1.20、0.60、0.30 GBq/L,该系列溶液分别与等体积载体溶液混合,即得到碘[131I]酸根化合物活度浓度为4.81、2.40、1.20、0.60、0.30、0.15 GBq/L的线性溶液。对溶液进行色谱分析,每种溶液重复测定2次,记录每个线性点的峰面积,计算其平均值,以碘[131I]酸根活度浓度对峰面积进行线性回归,线性曲线示于图3。由图3可知:碘[131I]酸根活度浓度在0.15~4.81 GBq/L具有良好的线性关系,线性回归方程为y=188.70x-39.97,线性相关系数r2=0.994。

图3 碘[131I]酸根的线性曲线
2.4.4检测限(LOD)与定量限(LOQ) 根据《中华人民共和国药典四部:9101分析方法验证指导原则》[10],HPLC可以直接采用信噪比(S/N)法确定检测限与定量限,检测限的S/N应≥3,定量限的S/N应≥10。根据已知低浓度溶液测出的信噪比,计算出能被可靠检测或者被可靠定量的碘[131I]化物溶液或者碘[131I]酸根溶液的活度浓度,即为二者的检测限或定量限。
(1) 碘[131I]化物
根据0.19 GBq/L线性溶液放射性主峰的S/N为34.35,推算出定量限与检测限适宜的活度浓度,分别为9.63×10-2GBq/L和3.21×10-2GBq/L。对其进行色谱分析,重复测定3次,记录S/N,结果列入表1和2。

表1 碘[131I]化物的检测限实验结果

表2 碘[131I]化物的定量限实验结果
(2) 碘[131I]酸根
根据0.15 GBq/L线性溶液放射性主峰的S/N为28.51,推算出定量限与检测限适宜的活度浓度,分别为7.91×10-2GBq/L和4.74×10-2GBq/L,对其进行色谱分析,重复测定3次,记录S/N,结果列入表3和4。

表3 碘[131I]酸根的检测限实验结果
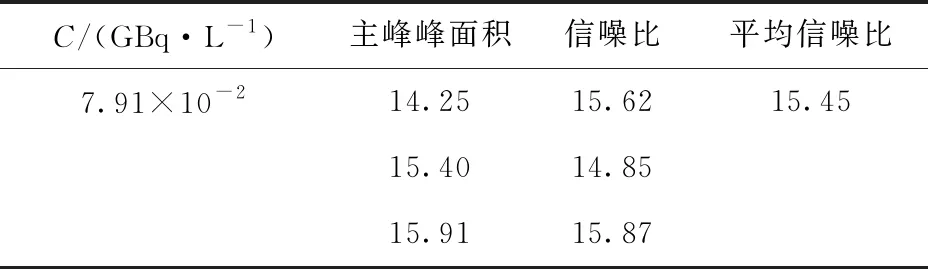
表4 碘[131I]酸根的定量限实验结果
2.4.5准确度 根据碘[131I]化钠胶囊的处方组成,取不含碘[131I]的空白辅料(抗氧剂A+填充剂B)于10 mL西林瓶中,将50 μL 活度为129.50 MBq碘[131I]溶液均匀滴在空白辅料上,即制备得到模拟胶囊内容物,放置10 min,使碘[131I]化物充分吸附,之后参照1.3.3节的方法制备成碘[131I]化物活度浓度为6.48 GBq/L(C理论)的样品溶液,按照上述过程平行制备6份样品溶液,对其进行色谱分析,记录碘[131I]化物放射性主峰峰面积,代入线性回归方程y=161.90x+466.32,计算得到碘[131I]化物活度浓度测定值(Cdet),结果列入表5。由表5可知:回收率在70.06%~75.31%,平均值为71.96%,相对标准偏差(sr)为3.38%。通过式(1)可计算出模拟胶囊内容物中碘化学含量(w)为40 μg/kg,满足待测成分含量对应的回收率(70%~125%)和重复性(sr=15%,n=6)的要求[10]。

表5 准确度实验结果
(1)
式中:A理论,碘[131I]的理论加入活度,MBq;a,碘[131I]的比活度,MBq/kg;m,模拟胶囊内容物质量,g。
2.4.6耐用性 主要考察了HPLC法色谱条件如色谱柱温度、流速、流动相pH值对测定结果的影响。本实验所用对照溶液1和对照溶液2按照1.3.1节的方法进行制备,待分析样品溶液参照2.4.2 节中测试溶液3的制备方法。对这两种溶液进行色谱分析,结果列入表6。由表6可知:对照溶液2中碘化物与碘酸根分离度(d)在7.37~12.88;待分析样品碘[131I]化钠放射性主峰的保留时间(tR2)与对照溶液1中碘化物峰的保留时间(tR1)的相对误差为0.44%~2.00%;待分析样品溶液中碘[131I]化钠的放化纯度按放射性峰面积归一化法计算均在97.0%左右,sr=0.10%(n=12)。以上结果均满足文献[10]中d≥1.5、tR2和tR1相对误差小于10%、放化纯度≥95%和sr≤2%(n=6)的要求。

表6 耐用性实验结果
2.4.7重复性 一名实验人员参照2.4.2 节测试溶液3的方法,制备某一活度浓度水平的6份样品溶液,活度浓度应在碘[131I]化物溶液的线性范围内(0.19~92.50 GBq/L)。对6份样品溶液进行色谱分析,并对溶液中碘[131I]化钠的放化纯度按放射性峰面积归一化法计算得到其平均值为97.12%,sr=0.31%(n=6),满足sr≤1%(n=6)的要求[10],表明该法重复性良好。
2.4.8中间精密度 两名实验人员按照测试溶液3的方法各制备同一活度浓度水平的6份样品溶液,活度浓度应在碘[131I]化物溶液的线性范围内。对12份样品溶液进行色谱分析,并计算样品溶液中碘[131I]化钠的放化纯度,得到放化纯度平均值为97.13%,sr=0.32%(n=12),满足sr≤2%(n=6)的要求[10],表明该法中间精密度良好。
2.5 最小进样量的确定
放射性流量检测仪对同一核素的检测效率不会随其化学形态不同而改变,在对碘[131I]离子和碘[131I]酸根均进行检测限与定量限的实验时,检测限分别为3.21×10-2GBq/L和4.74×10-2GBq/L,对应的S/N分别为5.39和8.27,二者活度浓度与S/N的比例关系分别为0.16和0.16,实验结果与理论一致。放射性化学杂质碘[131I]酸根的检测限(LOD)为4.74×10-2GBq/L和百分比(Y′)应小于5%(治疗用碘[131I]化钠胶囊放化纯度应不小于95%),最小进样量活度(Amin)由式(2)计算为1.90×10-2MBq,可确保放射性化学杂质百分比不小于5%时能被检测到。
Amin=LOD×0.02/0.05
(2)
式中:0.02为进样体积,mL;0.05为杂质碘[131I]酸根放射性量不应超过总放射性量的百分比。
2.6 不同活度浓度进样量下放化纯度的测定
取1粒胶囊内容物溶解于10 mL水中,超声助溶,然后静置,取上层清液,备用。加入一定量碘[131I]酸根溶液,混匀,得到活度浓度为103.60 GBq/L的溶液,取适量与载体溶液混合后活度浓度为51.80 GBq/L,依次稀释获得10.36、5.18、3.45、1.15 GBq/L的溶液,以上溶液均按测试方法进行分析,进样量为20.0 μL,按放射性活度计算进样量分别为1.04、0.21、0.10、6.90×10-2、2.30×10-2MBq。测得样品放化纯度均约在95.0%~95.5%,平均值为95.31%,sr=0.20%(n=5)。结果表明:在满足最小进样量和活度浓度线性范围的前提下,不同进样量对样品放化纯度检测结果没有影响。
3 结 论
建立了治疗用碘[131I]化钠胶囊放化纯度的Radio-HPLC测定方法,该法具有较强的专属性、较高的准确度和良好的重复性与中间精密度,可用于治疗用碘[131I]化钠胶囊放化纯度的质量控制,并为其它种类的放射性药品放化纯度Radio-HPLC测定方法的建立以及方法学验证提供参考。
猜你喜欢
钢铁钒钛(2022年4期)2022-09-19
农村青少年科学探究(2022年4期)2022-07-29
中国药业(2022年7期)2022-04-20
科学技术与工程(2021年31期)2021-11-23
食品安全导刊(2021年20期)2021-08-30
粉末冶金技术(2021年3期)2021-07-28
现代临床医学(2021年3期)2021-07-16
食品界(2018年8期)2018-09-03
中学生数理化·高二版(2016年6期)2016-05-14
