
相转移法制备PdMo催化剂及其氧还原活性
2021-11-04 13:54金碧瑶赵一鸣赵莲花
无机化学学报 2021年10期
刘 汝 金碧瑶 赵一鸣 赵莲花
(延边大学理学院,延吉 133002)
0 引 言
为了减少对传统化石能源的依赖,满足人们日益增加的能源需求,解决环境污染问题,人们研究了多种绿色能源,并研究了相应的能量转换装置。其中,燃料电池能够高效地将化学能转化为电能,且能量转化率高、环境友好,在电动汽车等电子设备上有广泛的用途,因而备受关注[1-4]。氧还原反应(ORR)在燃料电池的阴极发挥着重要作用,但是由于在碱性介质中阴极ORR的动力学滞后,其反应速度缓慢,需要大量的催化剂[5-7]。目前,Pt/C为常用的ORR阴极催化剂。但是,Pt的成本较高、地壳储量低且稳定性差(由奥斯瓦尔德熟化效应导致)[8-10],限制了燃料电池的商业化发展,因此需要开发一种非铂催化剂。
Pd不仅具有可与之媲美的活性,而且地壳储量相对较高,是最有希望替代Pt的金属。事实上,Pd作为ORR催化剂的研究已经取得了一定成果,例如:Shen等利用硅纳米线和氢氟酸在F、N共掺杂的石墨烯上制备Pd纳米粒子,该材料在0.1mol·L-1KOH中得到了优于商业Pt/C的半波电位[11]。另一项研究表明,于适当的热处理条件下合成的PdAu纳米链具有较好的ORR性能[12]。但是,由于钯与氧的结合能比较高,反应含氧中间产物难以脱附,催化剂容易中毒,影响其稳定性[13-14]。
为了提高Pd的催化活性,可以采用适当的方法对其进行调节。目前,增强催化活性的策略主要包括2大类:一是增加材料的活性位点,如结构纳米化、调控催化剂结构以暴露更多活性位点、暴露不同的晶面、调节催化剂尺寸和形貌[15-16]、制备负载型催化剂等;二是提高催化剂的本征活性,如合金化[17]、构筑核壳结构、加入功能载体[18]等。最近,严等[19]开发了一种简便的方法来制备Pd纳米立方体@Mo核壳结构材料作为高性能ORR催化剂,通过加入第二组元并形成核壳结构赋予了Pd-Mo协同效应,Mo涂层与Pd核存在一种强烈的相互作用,从而调节了Pd的几何结构与电子结构,使Pd的晶格发生收缩,电子发生从Pd到Mo的转移。除了核壳结构,引入其他金属与Pd形成合金也对Pd的电子结构和几何结构有很好的调节作用。罗等[20]报道,在W、Cu、Mo、Ni等众多金属中,Mo对Pd氧结合能的调节最接近最优值。其原因可能是Mo的电负性和半径与Pd比较相近,容易形成合金,可以通过配体效应和应力作用来调节Pd的电子结构和几何结构,优化氧结合能,提高催化活性。此外,不同尺寸的纳米粒子暴露的活性位点不同,活性也各不相同。如Jiang等[16]制备了3~16.7 nm的Pd纳米粒子,发现纳米粒子表面吸附的羟基随其尺寸的增大而增多;当尺寸为3~5 nm时,ORR活性随其尺寸的增大而增强;当尺寸大于5 nm时,表面过多的羟基堵塞活性位点,使得ORR活性随其尺寸的增大而降低。可见,催化剂的活性与颗粒尺寸是密切相关的。
催化剂的结构与其制备方法相关。目前大多数的Pd基催化剂采用水相法[21-22]合成,而在水相中制备的催化剂极性大、易团聚,使得纳米粒子的分散性差,活性位难以暴露在两相界面,影响其活性。而在有机相中纳米粒子的表面能更低,这使得其尺寸可控,从而具有更好的分散性,在催化领域的优势显著。但是大部分Pd以及其他金属盐前驱体是不溶于有机相的,因此其在有机相中的合成成为了难题。相转移法可以将不溶于有机溶剂的金属盐前驱体从水相转移到有机相中,在有机相中进行还原,从而增强纳米粒子的分散性,提高其反应活性[23]。
我们首次采用四丁基氢氧化铵(TBAOH)作为相转移剂,利用相转移法在二氯甲烷中制备了PdxMo/C(Pd/Mo的原子比x=1、2、3、4、5)纳米粒子。通过透射电镜(TEM)、X射线衍射(XRD)、X射线光电子能谱(XPS)分别对催化剂的形貌、几何结构和表面电子性质进行分析。并通过电化学测试分析了不同比例PdxMo/C催化剂的ORR活性,从而探究了其在碱性燃料电池中的应用。
1 实验部分
1.1 化学药品及试剂
PdCl2(无水粉末,99.999% )、MoCl5(无水粉末,99.6% )、NaBH4(粉末,98% )、TBAOH(10%,水溶液)均购于阿拉丁试剂(上海),CH2Cl2(AR)购自天津市科密欧化学试剂有限公司,Vulcan XC-72活性炭购自美国Cabot公司。金属负载量(质量分数)为20% 的Pt/C购自Alfa Aesar试剂。以上试剂在使用时未经进一步处理。
1.2 材料的制备
用相转移法合成金属负载量(质量分数)为20% 的PdxMo/C纳米粒子[23]。在不同比例的PdCl2和MoCl5前驱体中加入几滴浓盐酸,随后溶于100mL的去离子水中,充分搅拌溶解后加入100mL二氯甲烷(CH2Cl2),由于金属前驱体在有机相中溶解度很低,此时金属离子在水相中。然后,向上述溶液中逐滴加入相转移剂TBAOH,并进行磁力搅拌直到金属离子完全转移到有机相。取有机相中溶液,加入在硝酸溶液(质量分数为69% 的浓HNO3和H2O体积比为1∶1,搅拌5 h)中预处理过的Vulcan XC-72活性炭,搅拌3 h。在饱和N2氛围中向悬浮液中逐滴加入NaBH4溶液,搅拌过夜,经抽滤、去离子水洗涤,40 ℃真空干燥得到PdxMo/C(x=1、2、3、4、5)催化剂。
1.3 物性表征
采用TEM(JEM-2100F)对样品的形貌、尺寸大小进行测试,操作电压为120~200 kV。用XRD(Max-C,Rigaku)分析催化剂的相结构和晶体结构,采用Cu靶Kα射线源,λ=0.154 059 nm,工作电流为100 mA,加速电压为40 kV,扫描速率为5(°)·min-1,扫描范围为 10°~90°。利用 XPS(Lb250 UK)对 Pd、Mo等元素的价态进行分析,X射线源为AlKα。
1.4 电化学表征
PdxMo/C的电化学性能在CHI-660E电化学工作站上进行,在一个三电极电解池中,使用由催化剂负载的玻碳旋转圆盘电极(GC-RDE,3mm,0.070 7 cm2)进行测试。以催化剂包覆的玻碳电极为工作电极,1 cm2的铂片作为对电极,Ag/AgCl电极作为参比电极,电解液为0.1mol·L-1KOH。所有涉及的电位均已换算成相对于可逆氢电极(RHE)电位的数值。通过线性扫描伏安(LSV)法确定样品的ORR活性。在LSV测试前于电解质中通入30min O2使其达到饱和状态,并且测试过程中O2贯穿全过程,扫描速率为 10mV·s-1,转速为 1 600 r·min-1。电化学测试均在室温下进行。
将4mg催化剂分散到包含0.98 mL异丙醇和20μL质量分数为0.5% 的Nafion溶液中,超声混合30min直至成为墨水,然后将10μL墨水分5次涂在玻碳电极上,在空气中干燥1 h备用。电子转移数通过Koutecky-Levich方程计算[24-25]:

其中j为测量电流,jk为RDE的动力学控制电流。jl为理论极限扩散电流,ω为角速度,n为电子转移数,F为法拉第常数(964 85C·mol-1),D为氧气分子的扩散率(pH=5~13 时值为 1.9×10-5cm2·s-1),v为溶液的动力学黏度(0.1mol·L-1KOH 中值为 0.01 cm2·s-1),c0为溶液中氧气的浓度(pH=5~12时值为1.2×10-3mol·cm-3)。
2 结果与讨论
2.1 Pdx M o/C(x=1、2、3、4、5)催化剂的形貌表征
图1a~1l显示了不同比例的PdxMo/C催化剂的形貌和相应的尺寸分布图(右侧)。如图1所示,在有机相中制备的一系列纳米颗粒均较均匀地分散在碳载体上,没有大面积团聚的现象。通过统计100个粒子可知,样品颗粒尺寸均较小,集中在2~4 nm,并且呈圆形。Pd/C、Pd5Mo/C、Pd4Mo/C、Pd3Mo/C、Pd2Mo/C、PdMo/C的平均粒径分别为2.97、3.25、3.96、3.39、3.04和3.17 nm。良好的分散性和较小且均匀的尺寸归因于在有机相中的合成,使得纳米粒子的表面能更低,从而团聚现象减少,同时相转移剂TBAOH具有两亲性基团,也可作为一种表面活性剂,阻止粒子的过度生长以防止聚集现象发生。
2.2 Pdx Mo/C(x=1、2、3、4、5)催化剂的结构
通过XRD对样品的结构进行研究。根据衍射峰最强的Pd(111)晶面衍射峰的位置和半峰宽信息,利用式3(布拉格方程)、式4(谢乐公式)及式5计算晶格间距、晶粒尺寸和晶格常数,结果列于表1。
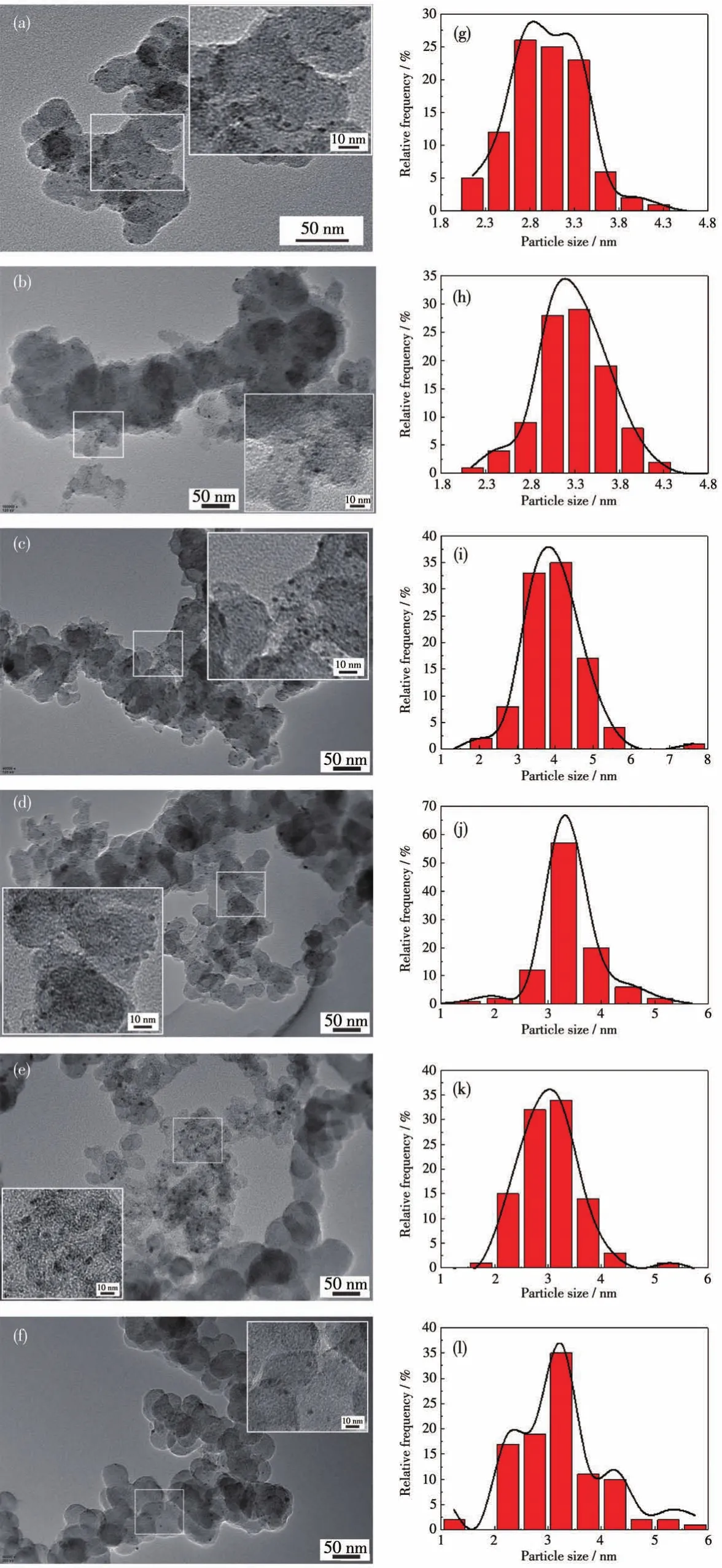
图1 (a)Pd/C、(b)Pd5Mo/C、(c)Pd4Mo/C、(d)Pd3Mo/C、(e)Pd2Mo/C和(f)PdMo/C的TEM图及局部放大图(插图)和(g~l)相应的尺寸分布图(右侧)Fig.1 TEM images and local magnification images(Inset)of(a)Pd/C,(b)Pd5Mo/C,(c)Pd4Mo/C,(d)Pd3Mo/C,(e)Pd2Mo/C,and(f)PdMo/C and(g~l)corresponding size distribution diagrams(right)
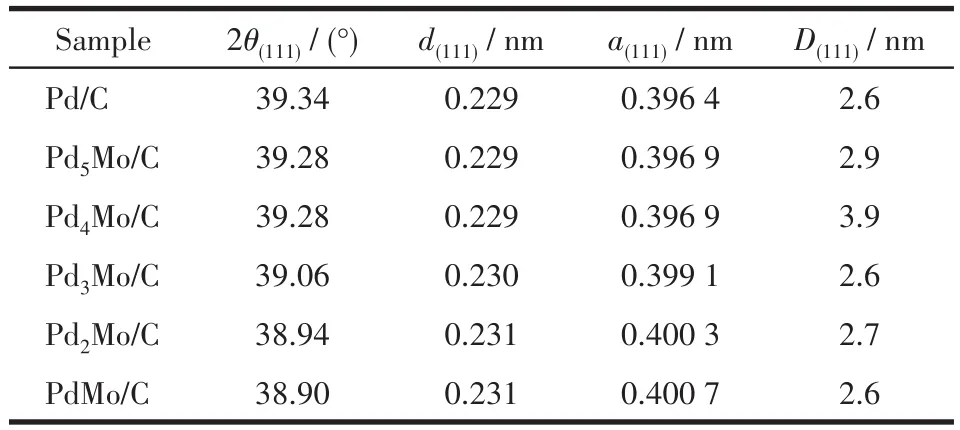
表1 Pd/C和Pdx M o/C(x=1、2、3、4、5)催化剂的晶格参数Table 1 Crystal lattice parameters of Pd/C and Pdx M o/C(x=1,2,3,4,5)
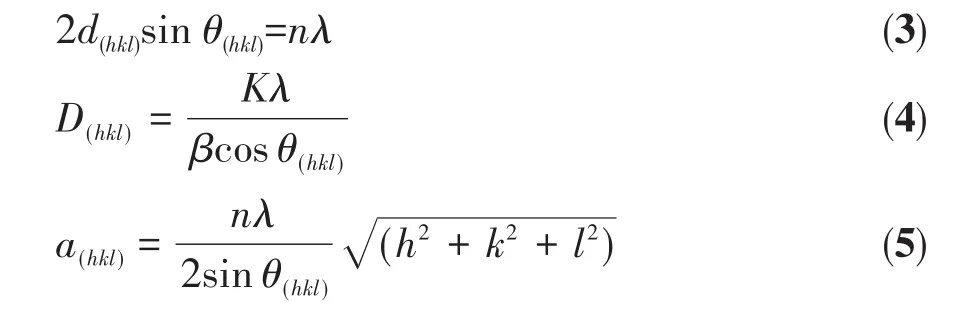
其中,h、k、l为衍射指标,d(hkl)为(hkl)晶面的晶格间距,n为衍射级数,λ为电子束轰击Cu靶产生的波长为0.154 nm的电磁波,θ为衍射角度,D(hkl)为平均晶粒度,K为Scherrer常数(0.920),β为所选峰的半峰宽,a(hkl)为晶格常数。
图2的XRD结果表明,用相转移法制备的Pd/C和PdxMo/C催化剂均为面心立方体(fcc)结构。在2θ=40.1°、46.7°、68.1°、82.1°处出现4个较强的衍射峰,分别归属于面心立方晶体的(111)、(200)、(220)和(311)晶面[20]。与Pd/C相比,随着Mo(原子半径更大)的加入,衍射峰发生不同程度的负移,对于加入较少量Mo的Pd5Mo/C和Pd4Mo/C而言,衍射峰的负移不明显,而对于Mo含量较大的Pd3Mo/C、Pd2Mo/C和PdMo/C而言,衍射峰随Mo的增加负移的更明显,且介于金属Pd与Mo的标准衍射峰之间,推测可能形成了合金。同时Mo的加入使PdxMo/C催化剂的晶格参数变大(表1),证明Mo修饰了Pd的几何结构。此外,当x=4时,样品峰型更加尖锐,证明晶体结晶性比较好。但是,从XRD图中并未观察到Mo的衍射峰,原因可能是Mo以非晶体形式存在或含量少。

图2 (a)Pd5Mo/C、(b)Pd4Mo/C、(c)Pd3Mo/C、(d)Pd2Mo/C、(e)PdMo/C和(f)Pd/C的XRD图Fig.2 XRD patterns of(a)Pd5Mo/C,(b)Pd4Mo/C,(c)Pd3Mo/C,(d)Pd2Mo/C,(e)PdMo/C,and(f)Pd/C
2.3 Pdx M o/C(x=1、2、3、4、5)催化剂的 ORR 性能表征
为了确定Mo的加入量对ORR催化活性的影响,测试了不同Mo含量的PdxMo/C催化剂的ORR活性,并与商业Pt/C进行比较。图3a是在O2饱和条件下、0.1mol·L-1KOH溶液中、1 600 r·min-1下催化剂的LSV曲线。如图所示,随着电势进行负向扫描,开始出现非零电流。通过曲线上最大斜率的切线与电流密度为0mA·cm-2的直线的交点横坐标来确定起始电位(Eonset),取极限扩散电流密度的二分之一所对应横坐标的电位来确定半波电位(E1/2),得到过电位的大小,进而评价ORR活性。由图可知,Mo含量不同,则起始电位不同,其由大到小的顺序为Pd4Mo/C(0.876 V)>Pt/C(0.870 V)>Pd3Mo/C(0.869 V)>Pd2Mo/C(0.868 V)>PdMo/C(0.864 V)>Pd5Mo/C(0.858 V)>Pd/C(0.848 V),在半波电位中也观察到类似的变化,即 Pd4Mo/C(0.813 V)>Pt/C(0.810 V)>Pd3Mo/C (0.809 V)>Pd2Mo/C (0.796 V)>PdMo/C(0.792 V)>Pd5Mo/C(0.786 V)>Pd/C(0.784 V)。

图3 (a)Pd/C、Pdx Mo/C和Pt/C的LSV曲线和(b)火山图Fig.3 (a)LSV curves of Pd/C,Pdx Mo/C and Pt/C,and(b)volcano plots
图3b显示了起始电位和半波电位的“火山型”趋势。不同Mo含量的PdxMo/C催化剂的ORR活性均高于Pd/C,说明Mo的加入对活性有较大的影响,且活性升高的幅度随Mo含量的增加呈先高后低的趋势,其中当x=4时,起始电位和半波电位均最大,并且高于商业Pt/C,但是,由于过量Mo的加入使得氧结合能增加,ORR活性减小,这种变化趋势可用Sabatier原理[26]来解释。
2.4 催化剂的电子转移数
为了在碱性电解质溶液中获得Pd4Mo/C催化剂上的ORR反应途径信息,采用RDE法研究了不同转速下的反应动力学。如图4所示(在O2饱和0.1mol·L-1KOH溶液中,扫描速率为10mV·s-1,扫速为400~2 500 r·min-1),ORR电流随转速的增大而增大,并通过式1和2中的Koutecky-Levich(K-L)方程计算电子转移数。应用不同转速下的电流密度和电势数据,在特定电势下(V=0.25、0.255、0.55 V)以ω-1/2对j-1作图得到直线,通过斜率得到电子转移数为3.85,说明在Pd4Mo/C催化剂上O2变为OH-的反应符合4电子途径。
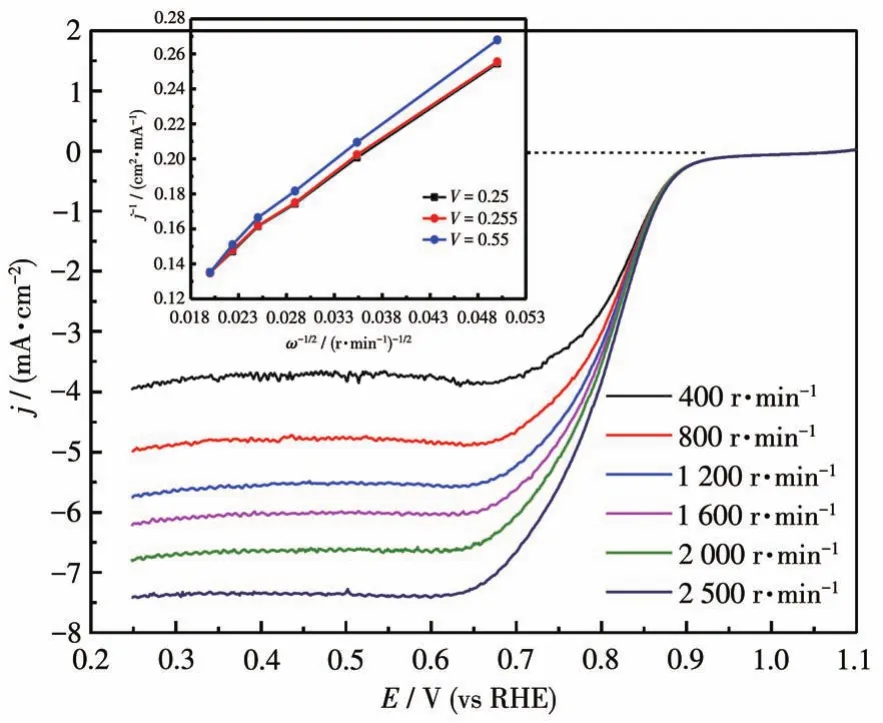
图4 Pd4Mo/C在不同转速下的LSV曲线和相应的K-L曲线(插图)Fig.4 LSV curves and K-L curves(Inset)of Pd4Mo/C with different speeds
2.5 催化剂的抗甲醇能力和稳定性
为了进一步了解催化剂PdxMo/C(x=1、2、3、4、5)在甲醇燃料电池中的潜在应用能力,在0.7 V(vs RHE)电压下,通过计时响应法在350 s时向O2饱和的0.1mol·L-1KOH溶液中加入3mL 3mol·L-1甲醇,评价催化剂的抗甲醇能力(图5)。由图可知,加入Mo之后催化剂的甲醇耐受性有明显的提高。PdxMo/C催化剂的性能衰减均小于Pd/C和Pt/C。其中,加入甲醇后Pd4Mo/C的电流保留率最高,经过1 000 s反应后仍然保留初始值的89.5%,证明Mo的引入可减缓CO在Pd表面的吸附,加速相应的氧化反应。
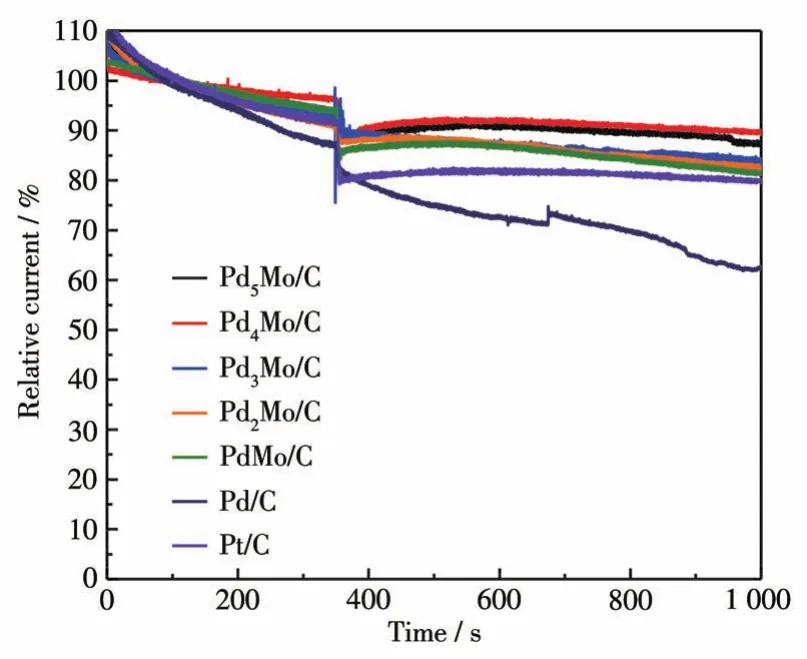
图5 加入3mL 3mol·L-1甲醇前后Pd/C、Pdx Mo/C和商用Pt/C的计时电流响应Fig.5 Chronoamperometric response of Pd/C,Pdx Mo/C,and commercial Pt/C before and after adding 3mL of3mol·L-1 methanol
图6是在0.7 V(vs RHE)下测定的计时安培响应曲线,可用于对比催化剂连续运行3 h的稳定性。由耐久性结果可以发现,不同Mo含量的PdxMo/C催化剂均有较好的稳定性,且高于Pd/C和商用Pt/C,其稳定性(电流保留率)由大到小的顺序为Pd4Mo/C(82.9% )>PdMo/C(81.5% )>Pd3Mo/C(78.1% )>Pd2Mo/C(65.6% )> Pd5Mo/C(60.1% )> Pd/C(47.1% )> Pt/C(40.6% )。同样,当x=4时,其稳定性最佳。其原因可能是由于Mo的加入调节了氧结合能,减缓了含氧物种的中毒,从而提高了催化剂的耐久性。同时,有机相中Pd前驱体的还原电位降低,晶体成核和生长过程变慢,得到的纳米粒子更加稳定。此外,预处理过的碳载体表面积大,表面含有丰富的含氧官能团,如羟基、羧基、羰基等,对金属纳米粒子具有较好的固定作用,对稳定性也起到积极的作用[27-28]。

图6 Pd/C、Pdx Mo/C和商用Pt/C的长期稳定性Fig.6 Long-term stability of Pd/C,Pdx Mo/C and commercial Pt/C
2.6 Pdx M o/C(x=1、2、3、4、5)催化剂的XPS表征
为了分析Pd4Mo/C催化剂对ORR电催化性能提高的原因,通过XPS对催化剂的电子结构进行了表征。图7a为Pd/C和Pd4Mo/C的XPS全谱图。如图所示,在Pd/C中检测出Pd、C、O的峰,在Pd4Mo/C中检测出Pd、Mo、C、O的峰,证明了Mo的存在。

图7 (a)Pd/C和Pd4Mo/C的XPS全谱图;(b)Pd/C和(c)Pd4Mo/C的Pd3d XPS谱图;(d)Pd4Mo/C的Mo3d XPS谱图;(e)Pd/C和(f)Pd4Mo/C的O1s XPS谱图Fig.7 (a)Full spectra of Pd/C and Pd4Mo/C;XPS spectra of Pd3d for(b)Pd/C and(c)Pd4Mo/C;(d)XPS spectra of Mo3d for Pd4Mo/C;XPS spectra of O1s for(e)Pd/C and(f)Pd4Mo/C
图7b和7c分别为Pd/C和Pd4Mo/CPd3d轨道的XPS拟合谱图。由图可知,在结合能约340.30和334.90 eV处的2个峰归属于金属Pd0,结合能在342.15和336.61 eV处的峰归属于Pd2+。说明在2种催化剂中均存在金属态的Pd和氧化态的Pd[29-30]。根据峰面积计算得到Pd/C中金属Pd0的含量为83.9%,而在Pd4Mo/C中金属Pd0的含量为84.5%,证明在加入电负性相对较小的Mo之后,Mo的电子可能向Pd转移,Pd得到电子使金属态的含量增大。Pd/C和Pd4Mo/C的Pd3d5/2的结合能分别位于335.38和334.88 eV。Pd4Mo/C的Pd3d结合能负移了0.50 eV,结合能的降低同样表明从Mo到Pd表面的电荷转移,说明加入的Mo与Pd之间发生了电子相互作用,这对Pd的电子结构产生了较大的影响,从而影响其电催化性能。
图7d为二者的Mo3dXPS拟合谱图[31]。结合能为235.60和233.51 eV、232.10和229.30 eV、227.90和226.50 eV的3对峰分别对应Mo6+、Mo4+、Mo0,说明Mo是以+6、+4和0价形式存在。
图7e和7f分别为二者的O1s的拟合谱图[32]。根据峰面积计算可知,Pd/C和Pd4Mo/C催化剂中羟基氧的含量分别为58.5% 和71.9%。显然,在Pd4Mo/C的表面吸附了更多的羟基。而根据TEM和XRD结果显示,x=4时催化剂的晶粒为3.9 nm,相对较大。根据Jiang等[16]报道,当Pd的尺寸在3~5 nm时,表面吸附的羟基随尺寸的增大而增多,这与XPS结果一致。催化剂表面羟基的吸附量大,能够为ORR提供更多的活性中间体,而在更小的颗粒上增加的羟基吸附可能堵塞ORR的活性位点,导致活性降低。因此,在不同比例的催化剂中,具有适当尺寸的Pd4Mo/C催化剂表面拥有更多的羟基氧,可能为催化反应提供更多的活性中间体,提高其ORR活性。
3 结 论
利用相转移法成功制备了分散性良好、粒径为2~4 nm的PdxMo/C纳米粒子电催化剂。在有机相中制备的粒子具有较好的分散性,有利于暴露更多的活性位点,当Pd、Mo原子比为4时,催化剂活性最佳。Mo的加入调节了Pd的几何结构和电子结构,随着Mo含量的增加,Pd的晶格参数变大,这对ORR性能有积极的影响;加入Mo之后Pd的结合能降低了0.50 eV,证明Mo和Pd存在电子的相互作用,电子从Mo向Pd转移,调节了Pd的电子结构。电化学测试结果表明,Pd4Mo/C催化剂具有良好的电化学活性,其起始电位(0.876 V)和半波电位(0.813 V)均高于市售Pt/C(0.870和0.810 V),电子转移遵循4电子途径。此外,与商业Pt/C相比,它具有长期耐久性和抗甲醇能力的优势。根据XPS对羟基氧含量的计算发现,适量尺寸的Pd、Mo原子比为4的催化剂表面吸附了更多的羟基,这可能为ORR反应提供了更多的活性中间体,从而有利于反应。本研究提供了一种合成PdxMo/C催化剂的新方法,作为一种高效催化剂,PdxMo/C有望应用于碱性燃料电池的阴极催化剂。
猜你喜欢
大学物理(2022年9期)2022-09-28
昆明医科大学学报(2022年1期)2022-02-28
新疆大学学报(自然科学版)(中英文)(2020年2期)2020-07-25
物理通报(2020年7期)2020-07-01
中成药(2018年2期)2018-05-09
浙江工业大学学报(2017年5期)2018-01-22
新乡学院学报(2016年6期)2016-12-01
中国塑料(2016年12期)2016-06-15
人间(2015年11期)2016-01-09
