
基于分子模拟预判Lyocell纤维原液着色体系中溶剂的稳定性
2021-11-03 01:40张玉梅王华平
纺织学报 2021年10期
靳 宏,张 玥,2,张玉梅,2,王华平,2
(1.东华大学 材料科学与工程学院,上海 201620;2.东华大学 纤维材料改性国家重点实验室,上海 201620)
Lyocell纤维因资源可再生和技术清洁化的优势,被誉为21世纪最具发展前景的绿色纤维[1]。随着Lyocell纤维规模生产技术的成熟,近年来产能增长迅速,功能化Lyocell纤维的研究开发是今后发展的必然趋势,抗原纤化、阻燃、抗菌、导电、荧光等功能性Lyocell纤维都有大量文献和专利公开报道[2]。与其他化学纤维一样,功能化Lyocell纤维的开发技术同样可以分为二大类:一类是成纤之后的功能处理,这类方法在原理和技术上与其他纤维素纤维基本一致,易于推广使用;另一类是纺前原液添加法,这类方法的实验室研究颇多,然而产品却极少,这在很大程度上取决于添加物对溶剂N-甲基吗啉-N-氧化物(NMMO)稳定性的影响程度。研究表明,NMMO在酸性、金属离子存在的情况下,其氮氧键易断裂并最终分解为吗啉和N-甲基吗啉,分解产物的生成会影响Lyocell纤维的溶解和纺丝[3],并使最终纤维性能和溶剂回收率下降[4]。许多功能性添加物的活性表面均可能与NMMO产生强相互作用而影响NMMO的稳定性[5]。
以有机或无机颜料作为添加剂制备原液着色Lyocell纤维,省去后续染色、干燥和废水处理等工序,具有节能、环保的优势,更符合Lyocell工艺绿色化的发展理念。以碳黑[6-7]、二氧化钛(TiO2)[8]、酞菁蓝[9]等作为着色剂对Lyocell纤维进行原液着色的研究,重点关注的是颜料表面改性对分散、着色溶液的可纺性、纤维结构性能等的影响;但对于着色剂是否影响溶剂NMMO稳定性的研究并不多见,除能够发生明显化学反应的金属离子类可以参考传统的化学分析法外,对于极性较强但发生化学反应需要时间和能量积累的体系,难以通过化学分析法预测规模化生产体系中因长期弱相互作用产生的潜在危害。前期本文课题组通过分子模拟的方法研究了纳米TiO2[10]、纳米碳黑[11]晶体表面与NMMO水溶液的相互作用,结果表明不同的晶体表面与NMMO、水分子的相互作用方式、作用能不同,从而影响纳米晶体在NMMO水溶液中的分散效果,且模拟计算结果与实验结果相吻合,也说明了分子模拟计算的可靠性。
用于原液着色的颜料很多,颜料因结构不同呈现出不同的颜色。从颜料结构角度,常用颜料可分为纳米碳材料、金属氧化物、有机颜料三大类。因颜料结构不同,与NMMO的相互作用也完全不同。为此,本文选择了6种具有代表性的纤维原液着色常用颜料进行研究,包括碳黑(CB)、二氧化钛(TiO2)、三氧化二铁(Fe2O3)、颜料红255(P.R.255)(3,6-二苯基-2,5-二氢吡咯并[3,4-c]吡咯-1,4-二酮)、酞菁铜、靛蒽醌。采用分子模拟方法分析不同颜料与NMMO分子的相互作用方式以及所引起的NMMO分子结构变化,判断颜料对溶剂NMMO分子稳定性的潜在影响;然后选取对NMMO分子影响较小的3种不同类型颜料,模拟其表面与NMMO水溶液间的界面相互作用,从而预测颜料颗粒在溶剂中的分散和团聚行为。
1 分子模拟计算方法
模拟计算软件为美国BIOVIA公司的Materials Studio 6.0,所用模块为Forcite,其中非键相互作用分为2部分:静电力采用Ewald[12]方法计算;范德华力采用Atom Based方法进行计算,截断半径为1.25 nm。温度控制采用Nose-Hoover方法[13]。
1.1 NMMO分子结构模型优化
按照文献[14-15]中的参数构建NMMO分子模型,初始结构为2个氧原子在对位的椅式构型(能量最低构型),然后对构建的NMMO分子初始模型进行几何结构优化。NMMO分子是由元素C、H、O和N组成的有机物,适合优化其几何结构的力场很多,但考虑到后续需要计算NMMO与各种颜料的相互作用,本文分别尝试了4种有一定普适性的力场,包括通用力场(Universal)、原子水平凝聚态优化力场(COMPASS)、Dreiding力场、聚合物一致性力场(pcff),分别进行几何优化和电荷分配,结果如图1所示。不同力场下优化的NMMO分子中原子点电荷和化学键参数计算结果不同,其中氧原子电负性直接影响到NMMO的物理和化学特性,所以氧原子点电荷大小是模拟中力场选取的重要参考因素[16]。

注:碳—灰色,氧—红色,氮—蓝色。图1 不同力场下NMMO分子的优化结果Fig.1 Optimization results of NMMO molecules under different force fields
考虑到COMPASS力场在金属氧化物固体模拟计算方面的优势,且NMMO分子在COMPASS力场下各共价键的键长及各原子的电荷分配与文献中更接近[15-16],统计了NMMO分子优化结果中部分原子点电荷及键长,并与相关文献对照,见表1、2所示。最终选择COMPASS力场作为NMMO分子结构优化和后续计算的力场。

表1 NMMO分子中各原子的电荷分配Tab.1 Charge of atoms in NMMO
表2 NMMO分子中各化学键键长Tab.2 Bond length in NMMO nm
1.2 NMMO水溶液结构模型优化
进一步在COMPASS力场下,对水分子进行几何结构优化,在此基础上构建了NMMO平均质量分数分别为10%、30%、50%和87%的NMMO水溶液模型,该模型具有三维的周期性边界条件,一般原胞模型的尺寸应大于力程的截断半径或体系中最大结构长度的2倍,因此,NMMO水溶液体系的原胞模型尺寸在3 nm以上,可以合理地计算镜像粒子间的作用[17]。接着对模型进行整体的几何结构优化,并在NPT系综(固定压强和温度)下进行了500 ps(在该模拟时间下,体系的密度达到平衡状态)的动力学模拟,结果如图2所示。

图2 不同质量分数NMMO水溶液模型的模拟结果Fig.2 Simulation results of different concentrations of aqueous NMMO solution models
统计了模拟结果中水分子和NMMO分子周围的氢键结合情况,如表3所示。可知:NMMO的平均质量分数在50%以下时,1个水分子平均可以与周围其他水分子形成1.5个氢键,1个NMMO分子平均可以与周围水分子形成2个氢键;而NMMO平均质量分数在50%以上时,水分子更多的与NMMO分子中的O1原子形成单独的氢键。氢键的限制条件为氢键的给体和受体氧原子间距离小于0.35 nm,氢键受体氧原子与氢原子的距离小于0.245 nm,氢键键角(H—Odonor—Oacceptor)小于30°。

表3 NMMO水溶液模拟结果中每个水分子和NMMO分子平均氢键数量Tab.3 Average number of hydrogen bonds of per water molecule and NMMO molecule in aqueous NMMO solutions models
1.3 界面相互作用模型的构建
选取TiO2、CB、Fe2O3、P.R.255、酞菁铜、靛蒽醌6种颜料为主要研究对象,并从晶体数据库中导入他们的晶体结构,在选定力场下进行几何结构优化。沿特定晶面切割每种晶体得到6种颜料的晶体表面,将优化后的NMMO分子置于6种颜料晶体表面上方,并在363 K下进行200 ps(总势能趋于平衡)的分子动力学模拟,初始的界面结构模型如图3(初始NMMO分子的方向为N—O键朝向颜料表面)所示。

图3 NMMO分子与颜料晶面间相互作用模型(初始结构)Fig.3 Interfacial model between NMMO molecules and pigment crystal planes (initial structure).(a)TiO2(110);(b)CB (001);(c)Fe2O3(010);(d)P.R.255 (110);(e)Copper (II)phthalocyanine (010);(f)Indoanthraquinone (010)
根据以上模拟结果,选取TiO2、CB、P.R.255这3种颜料表面构建了颜料表面/50%NMMO水溶液界面模型,并在363 K下进行1 000 ps(总势能趋于平衡)的分子动力学模拟,来研究实际溶液中颜料颗粒表面与50%NMMO水溶液的相互作用,模拟结果如图4所示。
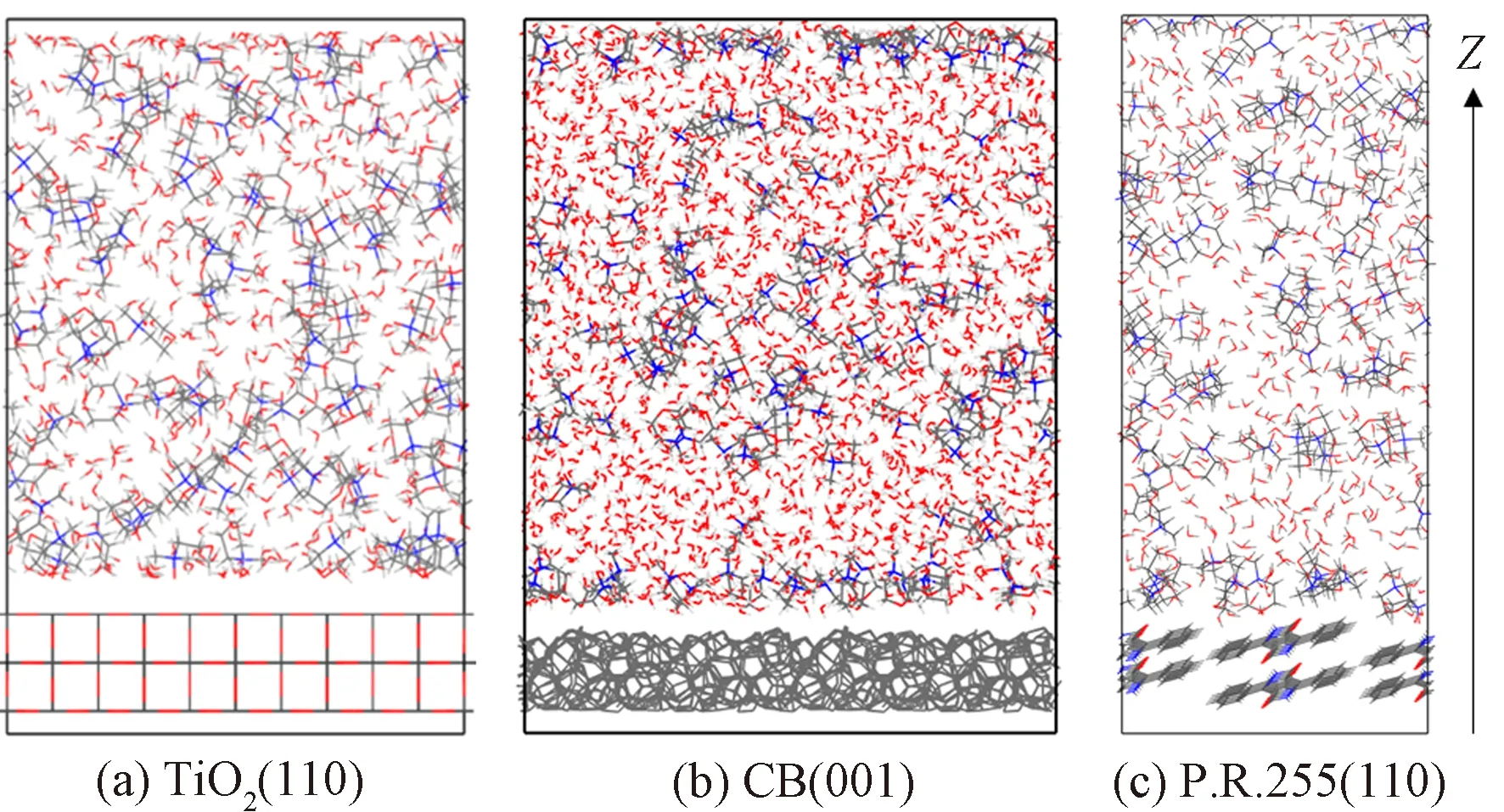
图4 50%NMMO水溶液/颜料晶面相互作用模型模拟结果Fig.4 Simulation results of interfacial models between aqueous NMMO solution and pigment crystal plane
2 结果与讨论
2.1 颜料对NMMO分子中N—O键影响
对363 K下的NMMO与颜料分子相互作用体系进行结构优化和动力学模拟,统计每一帧结果中N—O键键长分布。N—O键是NMMO分子中极性最强、最不稳定的键,键能约为125.5 kJ/mol,稳定状态下键长在0.130~0.145 nm之间伸缩,平衡键长为0.137 4 nm。N—O键平衡键长超过0.139 nm后就会变得不稳定[15],存在断裂的可能,因此可以用N—O键长的变化来判断颜料表面对NMMO分子稳定性的影响。此外还统计了C—N键键长分布作为对比。
模拟了在363 K下,真空中NMMO分子的振动,其中N—O键伸缩范围为0.130~0.145 nm,平衡键长为0.137 4 nm,如图5所示,符合文献[16]中报道的0.137~0.139 nm的稳定的N—O键键长范围。不同颜料表面上方NMMO分子中C—N键平衡键长如表4所示。

图5 NMMO分子在363 K下的N—O键键长分布Fig.5 N—O bond length distribution of NMMO at 363 K

表4 不同颜料表面对NMMO分子中C—N键键长的影响Tab.4 Influence of different pigment molecules on C—N bond length of NMMO molecule
进一步统计颜料晶体表面上方NMMO分子中的N—O键键长,并作其分布图,如图6所示。可知:TiO2、CB、P.R.255、靛蒽醌4种颜料表面对NMMO分子中N—O键键长影响较小,平衡键长在0.137 3~0.138 3 nm之间,说明这些颜料对NMMO分子的化学结构影响相对较小;而Fe2O3及酞菁铜对NMMO分子中N—O键键长影响较大,平衡键长在0.139 6~0.142 4 nm之间,易引起N—O键键长断裂,资料中相关的实验研究也表明铜离子和铁离子对NMMO的化学稳定性影响明显,会造成NMMO的分解[3-5],这也证明了模拟结果的可靠性。

注:统计分子数为50个;温度为363 K。图6 不同颜料晶体表面对NMMO分子中N—O键键长分布的影响Fig.6 Influence of different pigment crystal surfaces on the distribution of N—O bond length in NMMO molecules.(a)TiO2;(b)CB;(c)Fe2O3;(d)P.R.255;(e)Copper (II)phthalocyanine;(f)Indoanthraquinone
2.2 颜料表面对NMMO分子构型的影响
NMMO分子的2种椅式构型能量差较小,在极性环境下不仅会导致N—O键键长的变化,还可能导致NMMO分子构型的变化,为此统计了模拟过程中NMMO分子中O—N—C键的键角变化来判断颜料表面对NMMO分子构型的影响。
统计了模拟过程中不同颜料表面上方NMMO分子O—N—C键键角的演变,结果如图7所示。结果表明,模拟过程中所有颜料表面上方的NMMO分子中,C—N键键长及O—N—C键键角均无大幅度变化,而是在一定范围内波动(C—N键平衡键长振动幅度为±0.2 nm,O—N—C键平衡键角振动幅度为±10°),这种程度的振动不会改变NMMO分子构型[14]。

图7 不同颜料晶体表面对NMMO分子中O—N—C键键角的影响Fig.7 Influence of different pigment crystal surfaces on evolution of O—N—C bond angle in NMMO molecules.(a)TiO2;(b)CB;(c)Fe2O3;(d)P.R.255;(e)Copper (II)phthalocyanine;(f)Indoanthraquinone
2.3 颜料表面对NMMO水溶液的影响
根据上述研究结果,选择了对NMMO分子结构影响最小的TiO2、CB和P.R.255 3种颜料进行下一步的与NMMO水溶液的界面相互作用的模拟。在Lyocell原液着色过程中,颜料颗粒在NMMO水溶液中的分散是必不可少的环节,且颜料颗粒的分散效果直接影响纺丝稳定性和原液着色Lyocell纤维的品质,NMMO水溶液/颜料表面界面相互作用的研究可以预测颜料/NMMO/水体系的稳定性。
对界面模型的模拟结果中各组分氧原子在z轴局部物质的量浓度分布进行统计,并重点分析了界面处NMMO分子和水分子的排布,氧原子浓度分布如图8所示。

图8 不同颜料表面/50%NMMO水溶液界面模拟结果中氧原子摩尔分布Fig.8 Oxygen concentration profile of the simulation result of different pigment crystal surfaces/50% aqueous NMMO solution
与颜料表面产生相互作用的分子会在颜料表面/NMMO水溶液界面层形成吸附分子层(双电层),且由于局部高浓度会在氧原子浓度分布图中形成尖峰,与颜料表面相互作用较弱的分子则无类似现象。由水分子的氧原子浓度分布可知,TiO2表面会吸附水分子形成吸附分子层,而CB和P.R.255表面处水分子浓度与体相溶液中相近。NMMO分子同时具有强极性弱极性2种不同的氧原子,因此,对亲水和疏水的颜料表面均有一定亲和力,但对比NMMO中O1原子(强极性)和O2原子(弱极性)峰位置可以判断,O2原子与3种颜料表面的相互作用概率更多,说明此时溶液中NMMO分子与颜料表面的相互作用方式主要为范德华力,同时O1原子较少地直接作用于颜料表面。在一定程度上,颜料颗粒表面与溶剂分子间的亲和力会影响颜料颗粒在溶剂中的分散效果和分散稳定性,NMMO分子则可以通过范德华力与颜料颗粒表面作用,同时强极性端与溶剂中的水分子形成氢键,起到分散剂的效果,促进颜料分散,图9示出CB/NMMO水溶液模拟结果中界面层结构局部放大图,可以更直观地表现上述分析结果。
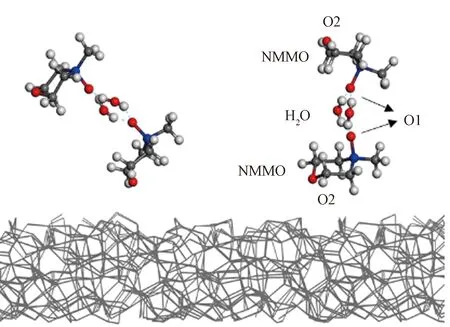
注:碳—灰色;氧—红色;氮—蓝色。图9 50%NMMO水溶液与CB表面之间的界面结构示意图Fig.9 Detailed structure of the interface between 50% aqueous NMMO solution and CB surface
3 结 论
通过分子模拟发现,不同颜料表面上方的NMMO分子的N—O键平衡键长在0.137 3~0.142 4 nm。根据N—O键键长的变化可以得出TiO2、CB、P.R.255、靛蒽醌4种颜料表面对NMMO分子中N—O键键长影响较小,而Fe2O3及酞菁铜对NMMO分子中N—O键键长影响较大,达到N—O键断裂的平衡长度极限(>0.139 nm),模拟计算表明铜离子和铁离子对NMMO的化学稳定性影响明显,会造成NMMO的分解。而N—O键键长超过后就会变得不稳定,趋于断裂。在保证NMMO分子稳定的前提下,NMMO水溶液中溶剂分子在颜料表面附近的排布仍会受物理相互作用的影响,并形成不同程度的双电层,进而影响颜料颗粒在溶剂中的分散和团聚行为。
猜你喜欢
椰城(2021年12期)2021-12-10
中学生数理化(高中版.高考理化)(2021年10期)2021-12-06
科教新报(2021年11期)2021-05-12
少儿科学周刊·少年版(2021年22期)2021-01-17
中学化学(2019年2期)2019-07-08
阅读(科学探秘)(2018年3期)2018-05-14
意林原创版(2017年11期)2017-12-01
广东教育·高中(2017年9期)2017-09-27
飞碟探索(2017年6期)2017-06-14
中学化学(2015年2期)2015-06-05
