
抑制肌球蛋白轻链激酶减轻大鼠压力超负荷诱导的心脏肥大*
2021-10-20 08:03周敬群
中国病理生理杂志 2021年9期
朱 丽, 周敬群△, 吴 钢, 王 顺, 毛 帅
(1三峡大学附属仁和医院,湖北宜昌 443001;2武汉大学人民医院心内科,湖北武汉 430060)
心脏肥大是心脏为适应高血压、主动脉瓣狭窄和心肌梗死等各种病理条件而增强心输出量的代偿过程,其特征是单个心肌细胞和整个器官的体积增大。最初,心脏肥大是一种保护心脏的代偿机制。然而,持续和过度应激诱发的病理性心肌肥大是不利的,可能最终导致心力衰竭、猝死和中风[1-2]。
肌球蛋白轻链2(myosin light chain 2,MLC2)是肌球蛋白的重要组成部分,其N 端有丝氨酸残基,可被肌球蛋白轻链激酶(myosin light chain kinase,MLCK)以Ca2+/钙调蛋白依赖/不依赖的方式特异性地磷酸化[3-6]。MLC2 的磷酸化在调控心肌收缩中发挥重要作用。近期通过对转基因、基因敲除和基因突变小鼠模型的研究表明,MLCK 和p-MLC2 在调节心脏肥大方面也至关重要。过表达MLCK 可以减轻压力超负荷诱导的心脏肥大,而敲除MLCK基因具有相反的作用,同时,MLC2 的磷酸化位点发生突变亦能加重压力超负荷诱导的心脏肥大[7-9]。另有研究表明,血管紧张素II(angiotensin II,Ang II)、苯肾上腺素和内皮素急性刺激心肌细胞和大鼠心脏均可导致MLCK、p-MLC2 和随后的肌节组织重组增加,而MLCK 抑制剂可阻止上述改变[10]。这些结果均提示MLCK 和磷酸化MLC2 可能成为干预心脏肥大发生和发展的重要靶点。
基于上述证据,本研究分别通过Ang II 刺激和腹主动脉缩窄(aorta banding,AB)建立心肌细胞肥大和大鼠压力超负荷心脏肥大模型,并通过抑制MLCK 来探讨干预MLCK 及p-MLC2 对心脏肥大的影响。
材 料 和 方 法
1 试剂与仪器
抗MLCK 和GAPDH 抗体(Abcam);抗MLC2、p-MLC2 和α-actinin 抗体(Cell Signaling Technology);MLCK 抑制剂ML-7(Santa Cruz Biotechnology);Trizol购自Invitrogen;cDNA 合成试剂盒和SYBR Green PCR Master Mix 购自Roche;RIPA 裂解液购自碧云天;BCA 蛋白定量试剂盒购自普利莱;PVDF 膜购自Millipore。心脏超声采用Vevo 2100 System(Visual-Sonics);显微镜购自Olympus;化学发光成像系统购自Bio-Rad。
2 实验动物
所有动物实验均经武汉大学人民医院动物伦理委员会批准,按照美国国立卫生研究院出版的第8版《实验动物管理与使用指南》(Guide NRC,2011)进行。本实验所用动物均购自武汉大学实验动物中心[许可证号:SCXK(鄂)2015-0027],包括1~3 d 的SD乳鼠和6 周龄的SD 大鼠。6 周龄雄性SD 大鼠体重180~220 g,饲养于武汉大学人民医院动物实验中心,饲养条件为温度20~25 ℃、湿度40%~70%和光照12/12 h光/暗周期。
3 方法
3.1 动物造模与MLCK 抑制剂处理 将60 只大鼠随机分为6组:(1)AB 0周组;(2)AB 2周组;(3)AB 4周组;(4)假手术组(sham 组);(5)AB 组;(6)AB+MLCK 抑制剂ML-7 组(AB+ML-7 组)。大鼠AB 手术诱导压力超负荷心脏肥大的过程简述如下[11-13]:首先,经腹腔注射水合氯醛(300 mg/kg)麻醉大鼠,褪去腹部鼠毛;然后,仰卧位固定大鼠,依次沿腹中线剪开皮肤、肌肉和腹膜,拨开肠道后分离腹主动脉,在肾上腺水平以7-0 丝线将22G 的钝性针头与腹主动脉一起结扎,闭塞腹主动脉,后迅速拔除针头,导致腹主动脉腔部分狭窄。对照组大鼠除未进行腹主动脉缩窄外,其余操作均相同。术后进行超声多普勒检测,以确定AB 手术是否成功。术后第1 天开始通过腹腔注射ML-7(1 mg/kg,溶于生理盐水,每天1次)对大鼠进行干预[14],另外两组大鼠通过腹腔注射接受相同体积的生理盐水,持续4周。
3.2 心脏超声检测与取材 如前所述,大鼠AB 术后4 周,通过超声心动图评估心脏功能[11-13]。首先,大鼠在仰卧位下以1.5~2%异氟烷进行吸入性维持麻醉。然后,采用Vevo 2100 System 经大鼠胸骨旁采集心脏短轴二尖瓣乳头肌切面的M 型超声心动图。根据美国超声心动图学会指南,测量至少3 个心动周期的左室收缩/舒张末期室间隔厚度(left ventricular end-systolic/diastolic interventricular septum thickness,IVSs/IVSd)、左室收缩/舒张末期后壁厚度(left ventricular end-systolic/diastolic posterior wall thickness,LVPWs/LVPWd)和左室收缩/舒张末期内径(left ventricular end-systolic/diastolic internal diameter,LVIDs/LVIDd),再取其平均值。计算左室收缩/舒张末期容积(left ventricular end-systolic/diastolic volume,LVESV/LVEDV)、左室射血分数(left ventricular ejection fraction,LVEF)和左室短轴缩短率(left ventricular fractional shortening,LVFS)[12-13]。
超声心动图完成后,立即称重并处死大鼠,以获取心脏、肺和胫骨。先将心脏浸入1 mol/L 氯化钾溶液中,使其停跳于舒张状态。然后,在0.9%生理盐水中清洗心脏、肺和胫骨,分别测量心脏和肺的重量以及胫骨的长度。然后,取部分心脏组织浸没于4%多聚甲醛中室温固定,其余心脏组织立即经液氮速冻后保存于-80 ℃冰箱用于RNA和蛋白提取。
另外,前3 组大鼠分别于AB 手术后0 周、2 周和4周取材,方法与上述一样。
3.3 病理学检测 心脏组织在4%多聚甲醛中室温固定24 h 后,采用石蜡包埋。将心脏组织切成5 μm的切片,随后分别进行HE 和Masson 染色并在显微镜下采集相应的图像。使用Image-Pro Plus 6.0(Media Cybernetics Inc.)对HE 和Masson 染色进行分析,分别用于测量心肌细胞的横截面积(cross-sectional area)和心肌纤维化程度。每组取不少于100 个心肌细胞计算心肌细胞的横截面积。每组至少测量50个左室间质和血管周围区域的视野,以确定心肌纤维化的程度。
3.4 心肌细胞培养和MLCK 抑制剂处理 原代乳鼠心肌细胞(neonatal rat cardiomyocytes,NRCMs)分离及培养的方法如前所述[12-13]。1~3 日龄的SD 乳鼠麻醉处死后迅速乙醇消毒胸部皮肤,剪开胸壁取出心脏置于PBS 中,洗净血液后修剪保留心室,剪碎至约1 mm×1 mm×1 mm 的组织小块。随后加入含0.08% II 型胶原酶和0.125%胰蛋白酶的消化液,37 ℃下消化每次10 min,第1 次弃上清,之后均收集上清,此过程持续至心脏组织被完全消化。随后通过差速贴壁的方法将心肌细胞与成纤维细胞分离,并以含10% FBS、1%青霉素/链霉素和0.1 mmo/L BrdU 的DMEM/F12 培养基培养心肌细胞。培养48 h后,将培养液换成1%的FBS。24 h 后,将心肌细胞随机分至3 个处理组:(1)PBS 组;(2)1 μmol/L Ang II组;(3)1 μmol/L Ang II+1 μmol/L ML-7组。处理48 h后收细胞[13,15]。细胞经免疫荧光双染(α-actinin 和cardiac troponin T)证实心肌细胞的纯度大于95%。
3.5 免疫荧光染色 心肌细胞免疫荧光染色的方法如前所述[11-13]。用于免疫荧光染色的心肌细胞培养于含有盖玻片的6孔板中,处理结束之后用PBS洗净,4%多聚甲醛室温下固定15 min,后以0.5% Triton X-100 室温下通透10 min,山羊血清室温下封闭30 min,随后与α-actinin 抗体(1∶100)4 ℃下孵育过夜。第2天与相应的Ⅱ抗在室温下孵育1 h。随后室温下DAPI 染色10 min。然后在荧光显微镜下采集心肌细胞荧光染色图片。采用Image-Pro Plus 6.0 软件测量心肌细胞表面积。每组测量超过100 个心肌细胞用以计算其表面积。
3.6 RT-qPCR 如前所述[12-13],根据制造商的说明,使用Trizol 从心肌细胞和冷冻大鼠心脏组织中提取总RNA,然后利用cDNA 合成试剂盒合成cDNA。然后,采用SYBR Green PCR Master Mix 进行荧光定量PCR,将内源性GAPDH 的mRNA 含量设为对照,量化目标基因的相对表达水平。用于扩增的心房钠尿肽(atrial natriuretic peptide,ANP)正向引物序列为5′-ATACAGTGCGGTGTCCAACA-3′,反 向 引 物 序 列为5′-AGCCCTCAGTTTGCTTTTCA-3′;脑钠肽(brain natriuretic peptide,BNP)的 正 向 引 物 序 列 为5′-CAGCTCTCAAAGGACCAAGG-3′,反向引物序列为5′-GCAGCTTGAACTATGTGCCA-3′;β-肌球蛋白重链(β-myosin heavy chain,β-MHC)的正向引物序列为5′-GCTCCTAAGTAATCTGTTTG-3′,反向引物序列为5′-AAGTGAGGGTGCGTGGAGCG-3′;GAPDH 的的正向引物序列为5′-ACAGCAACAGGGTGGTGGAC-3′,反 向 引 物 序 列 为5′-TTTGAGGGTGCAGCGAACTT-3′。
3.7 Western blot 实验[12-13,16-17]用RIPA 裂解液从心肌细胞和冷冻大鼠心脏组织中提取蛋白质,形成蛋白溶液。使用BCA 蛋白定量试剂盒测定蛋白浓度。通过SDS-PAGE 对每个样品中等量的蛋白质(20 μg)进行分离,然后转移到PVDF 膜上。在室温下用5%脱脂奶粉封闭这些膜1 h后,将这些膜与以下相对应的Ⅰ抗在4 ℃下孵育过夜:MLCK(1∶1 000)、p-MLC2(1∶1 000)、MLC2(1∶500)和GAPDH(1∶2 000)。然后,在室温下与Ⅰ抗相应的辣根过氧化物酶结合Ⅱ抗孵育1 h。后使用化学发光显影液在化学发光成像系统上曝光显示目标条带,并将目标条带用ImageJ(National Institutes of Health)进行量化分析。以每个样本中的GAPDH 蛋白含量作为内参照进行标准化,得到每个样本中目的蛋白的相对表达水平。
4 统计学处理
采用SPSS 20 进行统计学分析。连续型变量均以均数±标准差(means±SD)表示。多组间均数比较采用单因素方差分析(one-way ANOVA)和Tukey 检验。以P<0.05为差异有统计学意义。
结 果
1 抑制MLCK减轻Ang II诱导的心肌细胞肥大
在Ang II 刺激48 h 后,心肌细胞中ANP、BNP 和β-MHC 的mRNA 相对表达水平显著升高,同时心肌细胞表面积也显著增大;与单纯Ang II 刺激相比,给予MLCK 抑制剂同时处理后,心肌细胞中ANP、BNP和β-MHC 的mRNA 相对表达水平显著下降,同时心肌细胞表面积也显著减小,见图1。这些结果表明抑制MLCK可减轻Ang II诱导的心肌细胞肥大。

Figure 1. Immunofluorescence staining of primary neonatal rat cardiomyocytes and relative mRNA expression levels of cardiac hypertrophy markers. A:representative images of cardiomyocyte immunofluorescence staining and the quantitative analysis of cardiomyocyte surface area;B,C and D:the relative mRNA expression levels of cardiomyocyte hypertrophy markers,ANP,BNP and β-MHC. Mean±SD. n=3.*P<0.05,**P<0.01 vs PBS group;#P<0.05,##P<0.01 vs Ang II group.图1 原代乳鼠心肌细胞免疫荧光染色和心脏肥大标志物的mRNA相对表达水平
2 抑制MLCK 减轻Ang II 诱导的MLC2 磷酸化水平升高
Ang II 处理心肌细胞48 h 后,MLCK 的表达水平和MLC2 的磷酸化水平均显著升高(P<0.05);与Ang II 处理相比,MLCK 抑制剂处理后心肌细胞中MLCK 的表达水平没有出现明显的变化,但MLC2 的磷酸化水平显著降低(P<0.01),见图2。这些结果表明抑制MLCK 可减轻Ang II 诱导的MLC2 磷酸化水平升高。
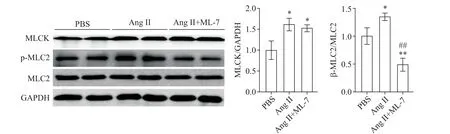
Figure 2. The protein levels of MLCK and phosphorylated MLC2 in cardiomyocytes after Ang II and MLCK inhibitor treatment for 48 h. Mean±SD. n=3.*P<0.05,**P<0.01 vs PBS group;##P<0.01 vs Ang II group.图2 Ang II和MLCK抑制剂处理48 h后心肌细胞MLCK和磷酸化MLC2蛋白水平的变化
3 MLCK 和磷酸化MLC2 蛋白水平在心脏肥大中先升高后降低
收集大鼠AB术后0周、2周和4周的心脏组织用于提取RNA 和蛋白,分别检测心脏肥大过程中肥大标志物的mRNA 表达水平以及MLCK 和磷酸化MLC2 的蛋白水平。随着大鼠AB 时间的延长,心脏肥大标志物ANP、BNP 和β-MHC 的相对mRNA 表达水平逐渐升高(P<0.05或P<0.01),见图3A。与0周组相比,AB 术后2 周大鼠心脏组织中MLCK 和磷酸化MLC2的蛋白水平显著升高,而AB术后4周MLCK和磷酸化MLC2 的蛋白水平显著下降,甚至低于0 周组,见图3B。这些结果表明在大鼠压力超负荷心脏肥大的过程中,MLCK 和磷酸化MLC2 的蛋白水平先升高后降低。

Figure 3. Relative levels of cardiac hypertrophy marker mRNA expression,MLCK protein expression and MLC2 phosphorylation after aorta banding for different time in rat heart tissues. A:the relative mRNA expression levels of cardiac hypertrophy markers ANP,BNP and β-MHC in rat heart tissues;B:the images of Western blot for determining the protein levels of MLCK,p-MLC2,MLC2 and GAPDH in rat heart tissues after aorta banding for 0,2 and 4 weeks,and the quantitative statistical results. Mean±SD. n=3.*P<0.05,**P<0.01 vs 0 week;#P<0.05,##P<0.01 vs 2 weeks.图3 大鼠腹主动脉缩窄不同时间后心脏肥大标志物mRNA表达、MLCK蛋白表达和MLC2磷酸化水平的比较
4 抑制MLCK改善压力超负荷大鼠的心功能
AB 术 后4 周,AB 组 大 鼠 的LVEDD、LVESD、LVEDV和LVESV均显著增加,而LVEF和LVFS显著降低,表明AB 组大鼠已出现明显的心功能不全并伴有严重的左室扩张;与AB 组相比,AB+ML-7 组大鼠的这些超声心动图参数有明显改善的趋势;sham 组和AB 组IVSd、IVSs、LVPWd和LVPWs的差异均无统计学显著性,但AB+ML-7组的上述值均显著大于AB组,见图4A、表1。这些结果表明抑制MLCK 改善了压力超负荷大鼠的心功能。

表1 大鼠腹主动脉缩窄及抑制MLCK 4周后的超声心动图结果Table 1. The results of echocardiography after 4 weeks of aorta banding and MLCK inhibition in rats(Mean±SD.n=10)
5 抑制MLCK减轻压力超负荷大鼠的心重指数
与sham 组相比,AB 组的心重/体重(heart weight/body weight,HW/BW)、心重/胫骨长(heart weight/tibia length,HW/TL)、肺重/体重(lung weight/body weight,LW/BW)和肺重/胫骨长(lung weight/tibia length,LW/TL)均显著增加;而AB+ML-7 组的这些指标均显著低于AB 组,见图4B~E。这些结果表明抑制MLCK减轻了AB导致的心重指数升高。
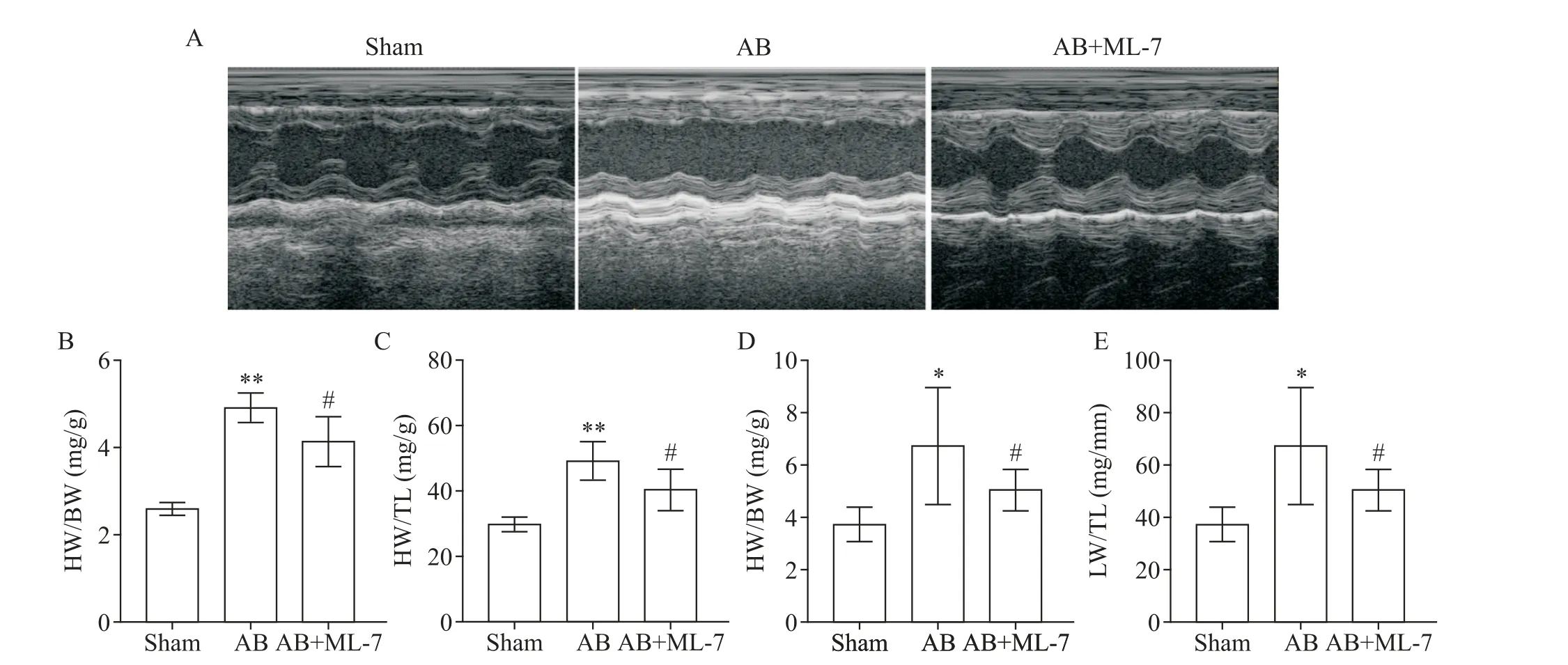
Figure 4. Echocardiography,heart weight(HW)indexes and lung weight(LW)indexese of the rats after aorta banding(AB)and MLCK inhibitor treatment for 4 weeks. A:the representative echocardiogram images in the rats after AB for 4 weeks;B and C:the HW to body weight(BW)ratio and HW to tibial length(TL)ratio of the rats;D and E:the LW to BW ratio and LW to TL ratio of the rats. Mean±SD. n=10.*P<0.05,**P<0.01 vs sham group;#P<0.05 vs AB group.图4 大鼠腹主动脉缩窄及抑制MLCK 4周后的超声心动图及心重和肺重指数的比较
6 抑制MLCK 减轻压力超负荷大鼠心脏组织病理学改变
心脏组织HE 和Masson 染色分别用于分析心肌细胞肥大和纤维化的程度。AB 组心肌细胞横截面积和纤维化比例均显著大于sham 组;与AB 组相比,AB+ML-7组心肌细胞的横截面积和纤维化比例显著降低,见图5A。这些结果表明抑制MLCK 改善了AB导致的心脏组织病理学改变。
7 抑制MLCK 降低压力超负荷大鼠心脏肥大标志物的表达
与sham 组相比,AB 组大鼠心脏肥大标志物ANP、BNP 和β-MHC 的相对mRNA 表达水平均显著升高,而这些标志物在AB+ML-7 组大鼠中均显著下降,见图5B~D。这些结果表明抑制MLCK减轻了AB导致的心脏肥大标志物表达升高。

Figure 5. HE and Masson staining and relative expression of hypertrophy markers in rat heart tissues. A:the representative images of HE and Masson staining,and the quantitative statistical results of cardiomyocyte cross-sectional area and fibrosis area(scale bar=50 μm);B,C and D:the relative mRNA expression levels of cardiac hypertrophy markers ANP,BNP and β-MHC in rat heart tissues. Mean±SD. n=10.**P<0.01 vs sham group;#P<0.05,##P<0.01 vs AB group.图5 大鼠心肌组织的HE和Masson染色以及肥厚标志物相对表达水平的比较
8 抑制MLCK 降低MLC2 的磷酸化水平而不影响MLCK表达
大鼠AB 术后4 周的心脏组织被用于检测MLCK表达和MLC2 磷酸化的水平。与sham 组相比,AB 组大鼠心脏组织中的MLCK 表达和MLC2 磷酸化水平显著降低;AB+ML-7 组大鼠的MLCK 表达水平出现了类似AB 组的下降,而MLC2 磷酸化水平显著低于AB 组,见图6。这些结果表明抑制MLCK 并不影响MLCK的表达,但影响MLC2的磷酸化。

Figure 6. The protein levels of MLCK and phosphorylated MLC2 in rat heart tissues after aorta banding and MLCK inhibition for 4 weeks were detected by Western blot. Mean±SD. n=10.*P<0.05,**P<0.01 vs sham group;##P<0.01 vs AB group.图6 大鼠腹主动脉缩窄及抑制MLCK 4周后心肌组织MLCK表达和MLC2磷酸化水平的比较
讨 论
MLC2是肌球蛋白的重要组成部分,可被其特异性的激酶MLCK 以Ca2+/钙调蛋白依赖/不依赖的方式激 活 而 发 生 磷 酸 化,生 成p-MLC2[4-6]。MLCK 和p-MLC2在调节心肌收缩、功能和疾病方面发挥直接且重要的作用[18-20]。尽管如此,目前并不清楚特异性地抑制MLCK 能否影响心脏肥大的病理进程。在本研究中,我们分别建立了Ang II 刺激产生的心肌细胞肥大和压力超负荷诱导的大鼠心脏肥大模型,并采用抑制剂对MLCK 进行特异性地抑制,结果表明:(1)抑制MLCK 可减轻Ang II 诱导的心肌细胞肥大;(2)抑制MLCK 可减轻Ang II 诱导的心肌细胞MLC2的磷酸化水平升高;(3)MLCK 表达和MLC2 磷酸化水平在大鼠心脏肥大中先升高后降低;(4)抑制MLCK 改善压力超负荷大鼠的心功能、心重指数、心肌细胞横截面积、心肌纤维化和心脏肥大标志物;(5)抑制MLCK 降低压力超负荷大鼠的MLC2磷酸化水平而不影响MLCK表达。
本研究通过Ang II 刺激诱导心肌细胞肥大,并检测心肌细胞表面积和肥大标志物以确定心肌细胞肥大模型成功建立。与PBS组相比,Ang II处理后心肌细胞表面积明显增大,且肥大标志物表达显著升高,而抑制MLCK 后上述改变明显减轻。另外,Ang II 处理后心肌细胞的MLCK 和p-MLC2 均明显升高,抑制MLCK 可减轻MLC2 的磷酸化而不影响MLCK的表达。上述结果证实MLCK 抑制剂ML-7 通过抑制MLCK 的催化功能进而发挥作用。同时早期的研究也表明,心脏肥大的激动剂包括Ang II、苯肾上腺素和内皮素急性刺激培养的心肌细胞和大鼠心脏可导致MLCK、p-MLC2和随后的肌节组织重组增加,而上述肥大刺激引起的肌节组织重组可被MLCK 的抑制剂阻止[10]。上述证据表明MLCK 和p-MLC2 参与并促进了心肌细胞肥大的发生。
超声心动图、组织病理学和RT-qPCR 的结果证实AB 手术可成功诱导大鼠心脏肥大。与sham 组相比,AB 引起LVEDD、LVESD、LVEDV 和LVESV 明显增加,同时LVEF 和LVFS 明显下降,而IVSd、IVSs、LVPWd和LVPWs没有明显的变化,这些结果提示大鼠已出现离心性和失代偿性心脏肥大;抑制MLCK后上述超声指标均得到改善,而IVSd、IVSs、LVPWd和LVPWs 均较AB 组增大,提示AB+ML-7 组大鼠仍处于向心性肥大向离心性肥大转变的阶段。
近期基于转基因、基因敲除和基因突变小鼠模型的研究普遍认为MLCK 和p-MLC2 在改善心脏肥大和心力衰竭的病理进程中发挥积极作用[7-9,21-23],而本项研究却通过抑制MLCK 使压力超负荷诱导的大鼠心脏肥大得到改善。之前的研究表明MLCK 和p-MLC2 在心脏肥大和心力衰竭中发生降低[9,24],但并未检测心脏肥大过程中MLCK 和p-MLC2 随时间的变化。本研究首次对心脏肥大过程中MLCK 和p-MLC2 随时间的变化进行检测,结果显示MLCK 和p-MLC2 在心脏肥大的早期升高,而在晚期降低。此外,4 周的游泳训练可增加野生型小鼠MLCK 和p-MLC2 的水平[9]。MLCK 和MLC2 磷酸化位点突变的小鼠在应对压力超负荷时表现出离心性心脏肥大,而非在野生型小鼠上观察到的典型向心性心脏肥大[8]。同时,胸主动脉缩窄1 周后可导致MLCK 和p-MLC2水平明显降低,紧随其后是代偿性心脏肥大发展为失代偿性心脏肥大[9],这些结果均提示MLCK 和p-MLC2 在心脏适应应激中发挥重要作用,在应对压力超负荷的早期甚至出现升高。而本研究也已证实MLCK 和p-MLC2 在压力超负荷心脏肥大的早期水平升高。而抑制MLCK 可改善压力超负荷引起的心脏肥大,可能与抑制心脏肥大早期MLCK 和p-MLC2的升高和功能,进而减轻心肌肌节重组和心肌重构相关。早期的研究也证实,即使在非病理状态下,过表达MLCK 也能够促进心肌细胞的肌节重组,而敲低MLCK仅能够轻微地减轻心肌细胞的肌节重组[5]。值得注意的是,MLCK敲除或MLC2 磷酸化位点突变的小鼠在基础状态下可自发出现心脏肥大,这一方面可能与基因敲除或突变影响了正常小鼠心脏的发育或生长有关;另一方面可能与正常的MLC2磷酸化平衡状态被打破,进而引起心肌异常做功有关[8-9]。另外,在压力超负荷心脏肥大的早期,抑制MLCK 改善心脏肥大的病理进程可能还涉及通过抑制MLCK磷酸化MLC2过程,进而发挥减少心肌收缩做功和降低心肌氧耗的作用。
本研究首次证明抑制MLCK 可改善压力超负荷诱导的大鼠心脏肥大,此作用可能涉及抑制MLCK功能及MLC2 磷酸化进而阻止心脏肥大早期肌节组织的重构。该发现为病理性心脏肥大的防治提供了新见解。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆明医科大学学报(2022年2期)2022-03-29
世界科学技术-中医药现代化(2021年7期)2021-11-04
心肺血管病杂志(2020年5期)2021-01-14
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
海南医学(2016年8期)2016-06-08
中国病理生理杂志(2015年8期)2015-12-21
安徽医科大学学报(2015年9期)2015-12-16
西南军医(2015年5期)2015-01-23
