
环青海湖4种生境土壤中原核微生物群落结构及分子网络特征
2021-10-12 05:31王宇姝盛海彦罗莎莎胡月明余玲玲
生态环境学报 2021年7期
王宇姝 ,盛海彦,罗莎莎,胡月明, ,余玲玲 *
1. 农业部华南热带农业环境重点实验室,广东 广州 510642;2. 华南农业大学资源环境学院,广东 广州 510642;3. 青海大学农牧学院,广西 西宁 810016;4. 广东省科学院生物工程研究所,广东 广州 510316;5. 广东省土地利用与整治重点实验室,广东 广州 510642
环青海湖地区位于中国青藏高原高寒地区农牧交错带,农业和畜牧业交叉分布,土地利用变化频繁,生态系统中的各类环境因素都处在边缘过度状态,属于生态系统脆弱区,尤其是农业和畜牧业的频繁交替过程会改变地表植被覆盖状况、土壤养分物质等环境因素,加剧土壤生态系统的退化(Liu et al.,2017;Wang et al.,2020)。改善土壤肥力和提高土壤质量成为这一区域发展的关键问题。其中,土壤微生物担当了包括土壤碳、氮循环在内的多个重要的生态系统功能,对维持生物多样性与生态系统功能和稳定性起着关键作用(Chapin et al.,2009),土壤微生物生态学特性是土壤质量和土壤生态系统功能的重要表征(Cruz et al.,2009;李娟等,2008)。高寒土壤微生物对气候、温度、水分等环境因子具有敏感的响应,不同生境土壤的空间异质性和环境因子的差异也会影响土壤微生物群落结构差异,并包含了大量未知和远古的微生物种群。有研究表明,高寒土壤原核微生物在不同生境存在不同的优势菌种,高寒草甸带土壤中细菌的优势菌门为放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和芽单胞菌门(Gemmatimonadetes)(朱平等,2017),在青藏高原冻融腹地不同生态系统中变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)及浮霉菌门(Planctomycetes)细菌为研究区优势菌种(张宝贵等,2012),北极冻土带的土壤中变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)和放线菌门(Actinobacteria)占主导地位(Koyama et al.,2014),在天山北坡高寒山地的研究中广古菌门(Euryarchaeota)和泉古菌门(Crenarchaeota)在古菌群落中占主导地位(吴尊凤等,2012)。
近年来高通量测序技术和分子网络分析技术的发展为研究土壤微生物的多样性、群落组成和与环境因子之间的相关性提供了有力的研究方法。分子生态网络分析技术能够更进一步地探究土壤微生物种间的相互作用,评估微生物群落结构变化引起的生态系统功能演变以及生态系统的稳定性(石文莉等,2018)。汪峰等(2014)通过构建土壤核心氮转化基因分子生态网络,表明了种植玉米作物显著改变了土壤氮转化基因网络中的关键基因;朱瑞芬等(2020)利用分子生态网络技术表明了在退化草地土壤中施氮会对土壤中的关键菌种产生影响,进一步可以通过合理施氮修复退化草地土壤;Zheng et al.(2017)比较了不同盐度土壤中原核微生物群落分子网络,发现高盐度胁迫加强了微生物间的相互作用来适应高盐度的生境。然而,针对高寒农牧交错区不同利用方式土壤中原核微生物的群落结构组成和菌种间相互作用的研究目前较少。本文选取环青海湖地区农牧交错带不同类型土壤原核微生物为研究对象,利用Illumina Miseq测序探究了不同土壤类型土壤原核微生物多样性、群落组成结构特征以及影响微生物群落的主要环境因子;利用随机矩阵原理构建了不同土壤利用类型土壤原核微生物分子生态网络,探究不同利用类型土壤原核微生物间的相互作用,解析土壤类型与原核生物群落的相互作用的关系。研究高寒生态系统中土壤微生物群落结构和分子网络特征能有效阐明土壤微生物对气候变化的响应进而表征当地土壤质量和环境变化,有助于推进环青海湖农牧交错带的可持续发展。
1 材料与方法
1.1 采样地点与方法
研究地位于青藏高原东北部,环青海湖进行采样,采样时间为2015年5月,期间以晴和多云天气为主。采样模式是选择了4种不同生境的样地进行采样,采样点按生境类型分为山地(XSo)、草地(DSo)、牧场(LSo)和农田(QSo)。其中,山地选择橡皮山生态保护区的冻土地带(36°45′15″N,99°36′21″E),植被主要以针茅和苔草为主;草地选择倒淌河牧场天然草地(36°24′10″N,100°58′35″E),植被是以芨芨草为主的禾本科、莎草科、杂类牧草为主;牧场选择青海湖近东岸草场(36°30′22″N,100°44′03″E),以禾本科牧草为主要植被,其次是莎草科、菊科、蓼科等;农田选择青海湖岸边以种植青稞为主的地块(36°33′24″N,100°31′58″E)。采用梅花布点法采集5个位置(区域内中心点及4个顶点)土壤表层(0—20 cm)的土壤样品,除去动植物残体及石块,混合均匀即为一个样品,每个采样点有设有3个重复,共12个样品用于后续分析。采集土壤样品后立即将土样放入便携式冰盒保存,随后,一部分土样存于−20 ℃冷冻冰箱用于土壤原核生物高通量测序,另一部分存于 4 ℃条件下保存用于土壤理化性质测定。
1.2 土壤理化性质测定
采用玻璃电极法测定土壤pH(土꞉水=1꞉2.5),重铬酸钾-外加热法测定土壤有机碳(SOC)含量,采用半微量凯氏定氮法测定总氮,钼锑比色法测定总磷,火焰光度法测定总钾,水蒸气蒸馏法测定有效氮,比色法测定有效磷,原子吸收法测定有效钾(鲍士旦,2000)。
1.3 高通量测序
土壤总DNA采用PowerSoil DNA Isolation Kit(MoBio,USA)试剂盒提取。提取方法按试剂盒说明书进行。采用 Nanodrop 2000(Thermo,UAS)测定提取后土壤DNA浓度及纯度。纯化、质控后,建立Illumina测序文库,采用Illumina MiSeq平台进行 Paired-end 250测序(美吉生物,上海,http://www.majorbio.com)。测序引物选取细菌16S rRNA V4—V5 区域的 338F(5′-ACTCCTACGGGA GGCAGCA-3′)/806R(5′-GGACTACHVGGGTWTC TAA-3′)和古菌16SrRNA V3-V5区域的Arch344F(5′-ACGGGGYGCAGCAGGCGCGA-3′)/Arch915R(5′-GTGCTCCCCCGCCAATTCCT-3′)。测序结果使用Flash软件和Trimmomatic软件拼接并去除低质量序列,根据序列两端的序列结构和引物序列得出有效序列,校正序列方向。使用UPARSE(version 7.1 http://drive5.com/uparse/)软件将相似度阈值97%以上的序列归为一个 Operation Taxonomic Unit(OTU),生成OTU表格,采用RDP classifier贝叶斯算法对OTU代表序列与数据库(采用16S rRNA基因序列数据库 Greengenes)进行比对,获得各个OTU的物种分类信息。使用MOTHUR(v.1.30.1)对OTU进行α多样性指数进行分析,利用R语言vegan包分析和作RDA图,Mantel test使用QIIME软件对环境因子与微生物群落的相关性进行分析。
1.4 网络构建与分析
对高通量测序获得的OTU数据进行标准化处理,在 Molecular Ecological Network Analyses Pipeline(MENA)网站上传数据后,构建Pearson相关性矩阵,基于随机矩阵理论(random matrix theory,RMT)通过设置合适的阈值,构建微生物分子生态网络,研究在不同生态系统下高寒土壤微生物群落之间的相互作用。利用Cytoscape 3.8.0软件对微生物分子生态网络进行可视化处理,得到各群落间网络结构。节点代表群落中的物种,节点间的连线表明了物种间的相互作用关系。连通度代表直接与某节点相连节点的数目,路径长度代表节点与节点之间的最短距离,聚类系数代表了一个节点与其他节点之间的连通程度,模块性表现了分子生态网络中的模块化特征。
1.5 统计分析
采用SPSS 21.0软件进行样地间土壤理化性质和土壤微生物群落丰度数据的单因素方差分析,采用Duncan进行差异显著性分析。利用Pearson相关性分析分子生态网络分别与环境因子之间的相关性。
2 结果与分析
2.1 土壤化学性质及养分状况
通过测定土壤的 pH、有机碳(SOC)、全氮(TN)、有效磷(Olsen-P)、碱解氮(AN)、速效钾(AK)和碳氮比来表征土壤的化学性质及养分状况。4种不同生境的土壤皆呈现碱性且存在显著差异(表 1),其中山地(XSo)的土壤呈弱碱性;而牧场(DSo)土壤的pH显著高于其他三者;SOC、TN和AN都表现为在山地土壤中显著高于其它3个样地,草场(LSo)、农田(QSo)次之,在草场土壤中含量最低;在农田中有效磷的含量最高,速效钾的含量则显著低于其他生境;山地土壤的速效钾和碳氮比较显著高于其他生境。
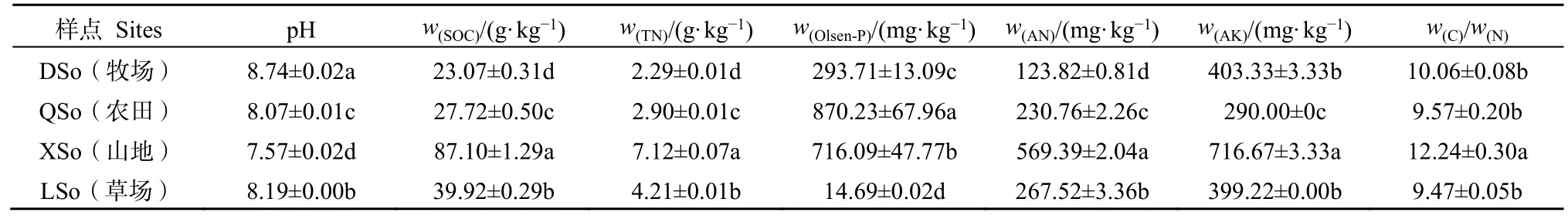
表1 不同生境土壤化学性质及养分状况Table 1 Soil chemical properties and nutrient status in different habitats
2.2 土壤微生物群落组成差异
采用Illumina MiSeq(PE250)对4种不同生境土壤测序分析,得到228833条细菌原始序列含有1432个OTU,古菌原始序列310644条含有309个 OTU。随着细菌和古菌测序量的升高,稀释曲线虽然还存在增加的趋势,但是增加速率趋近于平缓,说明测序数据量达到了可分析水平(图1)。
图1 稀释曲线图Fig. 1 Rarefaction curve
α多样性表征了微生物群落的丰富度和多样性,采用Shannon指数和Chao1指数对群落的α多样性进行评价。QSo的细菌丰富度和多样性最高且与其他生境差异显著(图2),DSo、XSo和LSo之间细菌丰富度没有显著差异;细菌Shannon指数LSo显著高于DSo和XSo,DSo和XSo之间没有显著差异。在4种不同生境的古菌多样性中,XSo的Chao1指数显著高于其他三者,说明在山地土壤中的古菌群落丰富度较高且显著高于其它生境。Shannon指数表明XSo的群落多样性最高,而DSo和QSo之间多样性没有显著差异,并低于XSo。在4种样地中,LSo的多样性最低且显著低于XSo。
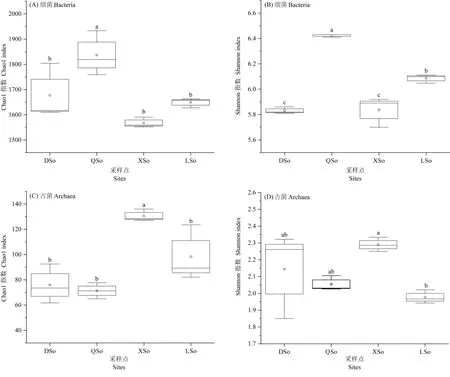
图2 不同生境土壤微生物α多样性Fig. 2 α-diversity of prokaryotic community in different soil habitats
对 4种不同生境土壤微生物群落在门水平上进行分布组成分析,采用线性判别分析(LDA)探究不同生境下土壤微生物群落存在差异的主要组分(图3)。由图3A可知,在DSo土壤中的关键细菌群落分别为 Proteobacteria、Actinobacteria、Acidobacteria、Chloroflexi和 Gemmatimonadetes,其中Actinobacteria所占比例在最大,达到29.61%;在 QSo土壤中细菌关键种共有 6门,其中Bacteroidetes是QSo土壤中的独有优势种,占比达到10.66%;在XSo和LSo中土壤优势菌种包含了Proteobacteria、Actinobacteria、Acidobacteria、Chloroflexi、Gemmatimonadetes和特有的Nitrospirae。由LDA分析结果(图3A)可以得出,在 DSo中土壤细菌群落差异主要来源于Actinobacteria、Gemmatimonadetes、Chloroflexi和Proteobacteria;QSo土壤中影响细菌群落差异的关键菌群为 Bacteroidetes和 Proteobacteria;而Nitrospirae、Acidobacteria和Proteobacteria是影响XSo土壤群落差异的主要组分;在 LSo中,Actinobacteria和Gemmatimonadetes作为关键菌种影响了细菌群落差异。
不同生境土壤中古菌的群落组成及其群落在门水平上差异的关键菌种(图3B)显示在DSo和XSo土壤中的优势古菌种为 Euryarchaeota和Thaumarchaeota,而在 QSo和 LSo中Thaumarchaeota是唯一优势菌种。LDA分析结果表明在DSo土壤中Euryarchaeota是决定古菌群落结构差异性的关键菌种;XSo中 Euryarchaeota和Thaumarchaeota共同影响了群落结构差异性;QSo和LSo中Thaumarchaeota是唯一优势菌种决定着不同生境群落间差异。

图3 不同生境土壤微生物群落组成与组间LDA分析结果分布Fig. 3 Soil prokaryotic community composition in different habitats and LDA analysis
2.3 环境因子对微生物群落的影响
基于RDA分析来解释环境因子对不同生境土壤微生物群落的影响。环境因子对土壤细菌群落的总解释量为74.60%,第一轴解释了48.56%,第二轴解释了26.04%(图4A)。DSo受pH影响显著,XSo则受到SOC、TN、AN、AK和碳氮比的影响。其中pH、SOC、TN、AN、AK和碳氮比都与土壤细菌群落显著相关(表2)。
环境因子解释了古菌群落的75.84%(图4B),第一轴和第二轴分别解释了51.37%和24.47%,pH主要影响DSo的土壤古菌群落,XSo主要受TN、AN、SOC、AK、碳氮比和Olsen-P的影响。环境因子中pH、SOC、TN、AN、AK和碳氮比显著影响了土壤古菌群落(表2)。
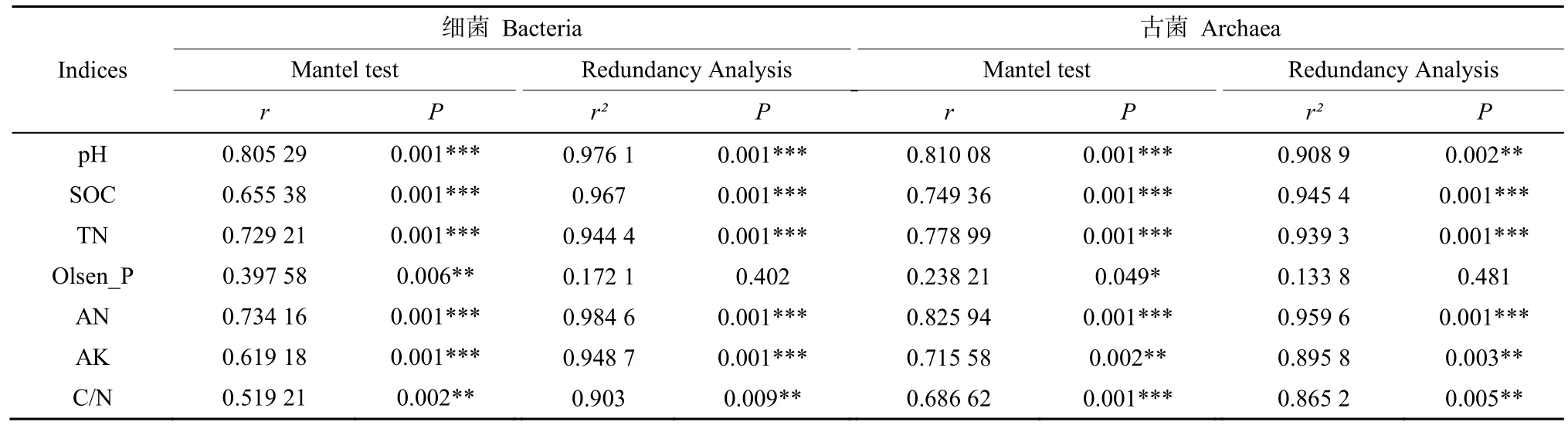
表2 土壤化学性质与微生物群落结构的Mantel检验和冗余分析Table 2 The Mantel test and Redundancy Analysis between soil chemical properties and nutrient status and microbial communities
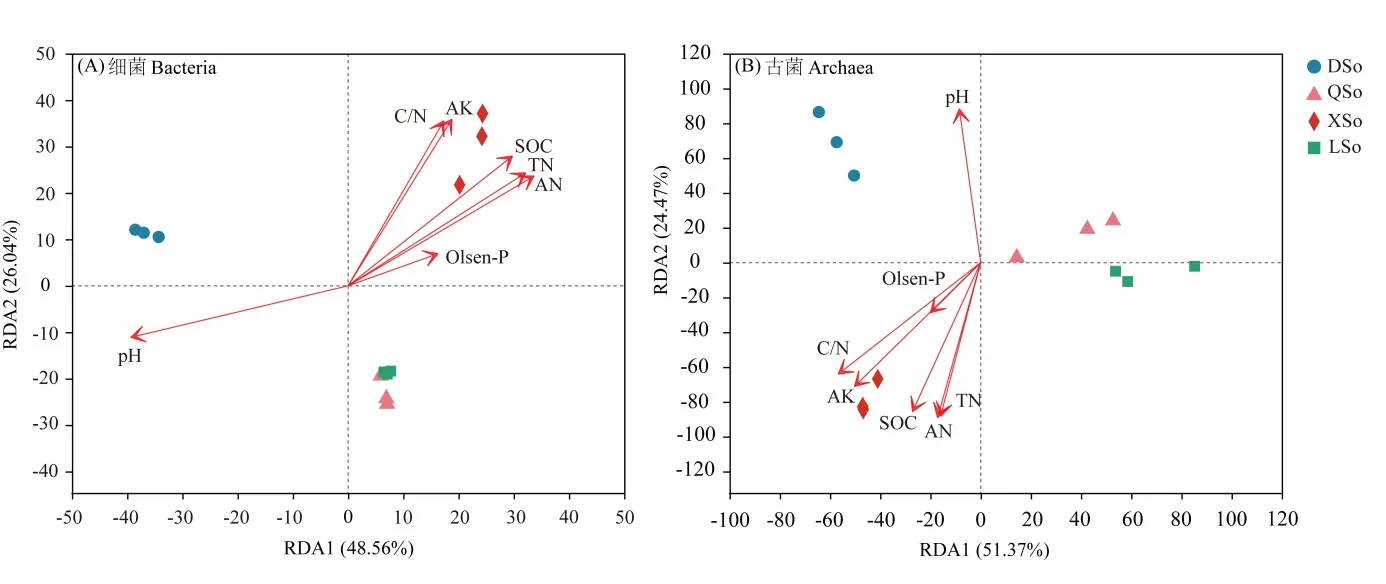
图4 不同生境土壤微生物群落与环境因子RDA分析Fig. 4 Redundancy analysis of soil microbial communities in different habitats
2.4 微生物分子生态网络分析
基于4种不同样地中细菌和古菌的OTU数据分别构建分子生态网络,网络中的节点代表群落中的物种,边代表物种间的联系。土壤细菌群落网络包含576个节点和2124条边组成(图5A,表3)。XSo土壤中细菌物种间连接数较多,相互作用关系更复杂,在网络中正相关占多数表明细菌物种间更多的是合作或共生关系;QSo网络中正相关占主导作用,负相关大多是来自硝化螺旋菌门;在 DSo土壤中细菌间的连接数减少,负相关也明显增多,表明在 DSo土壤中细菌物种间的相互作用较少且竞争关系较强;LSo网络中存在唯一菌种Tectomicrobia。
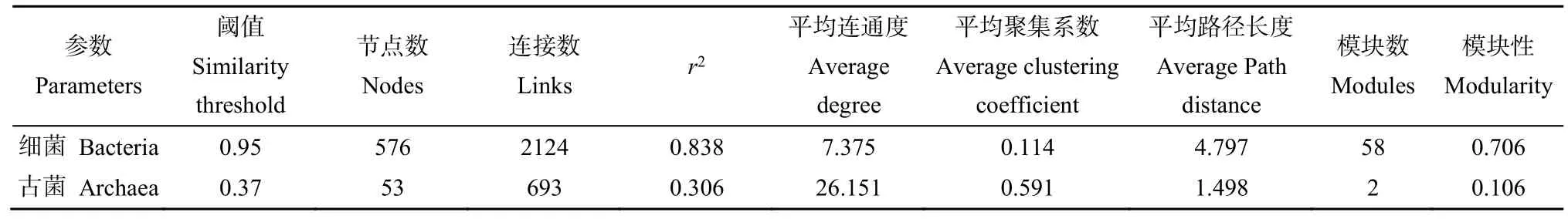
表3 微生物分子生态网络特征参数Table 3 Characteristic parameters of the molecular ecological network of soil microorganism
土壤古菌群落网络包含53个节点和693条边(图5B,表3),Thaumarchaeota在4种不同生境土壤中均存在,在古菌群落中具有重要位置。在4种生境中 XSo网络节点数和连接数较多,表明物种间相互作用强,种间关系复杂;QSo网络中负相关占大多数,种间竞争关系较强;DSo中网络连接和节点数较少。对比不同生境土壤细菌和古菌网络属性,细菌网络的节点数和连接数更多,节点间的联系更加复杂;而古菌网络的平均路径长较小,平均连通度和聚类系数较高,说明古菌种间的物质信息传递效率更高,对外界有敏感的响应,但是群落结构更加脆弱。

图5 不同生境土壤微生物菌群间的网络相互作用Fig. 5 Ecological networks of soil microbial community in different habitats
3 讨论
高寒生态系统下土壤微生物对全球气候变化响应敏感,气候和环境条件的改变可以影响微生物群落组成和功能,进而影响高寒生态的系统功能(褚海燕,2013)。对具有高海拔、强光照、气温差异大等特点的环青海湖农牧交错区,土壤利用类型一直被认为是微生物群落结构的主要决定因子(李冰等,2020)。农牧交错区土地的利用方式会对土壤碳氮库造成改变,进而影响土壤微生物群落结构的组成和功能(Bossuyt et al.,2001),针对该地区土壤微生物群落结构的研究有助于揭示高寒生态系统下土壤质量和利用效率。本研究对环青海湖地区 4种不同土地利用方式下土壤原核生物进行研究,阐明高寒地区不同生境下土壤原核生物群落对土地利用方式的响应过程与特点,不同生境的土壤由于人类干扰和植物覆盖不同等方面差异可能导致土壤化学性质和养分情况存在一定的差异。在环青海湖农牧交错区,4种不同生境(农田,牧场,草地和山地)土壤的化学性质及养分状况存在显著差异,不同生境土壤化学性质和养分状况不同使得微生物群落结构和稳定性也存在一定的差异。本研究显示,土壤中的SOC、TN、AN、AK都表现为在XSo土壤中含量最高,由于人为干扰,在土壤中施肥导致土壤矿化,全氮含量减少,同时外源养分也影响了Olsen-P的含量;农田收获和牧场放牧可能导致土壤中的有机碳含量降低。同时,土壤各项化学性质和养分指标也受到植株生长和气候条件等方面的显著影响(孙小丽等,2015)。
在农田生态系统中,合理施肥可以促进土壤有机碳、氮的积累,并驱动着农田土壤微生物群落结构演变(Cruz et al.,2009),在本研究中,农田(QSo)土壤细菌的丰富度和多样性最高且达到显著水平,可能是农田土壤的外源性养分输入影响了土壤理化性质和植物生长发育,进而改变了土壤细菌多样性(Wei et al.,2008)。土壤古菌的Chao1指数在山地(XSo)土壤中显著增加,而Shannon指数在草地(DSo)、农田(QSo)和山地(XSo)中较高,仅在山地(XSo)Shannon指数达到了显著较高水平。这可能由于山地(XSo)土壤中的可利用的SOC显著较高,增加了土壤中古菌群落的丰富度和多样性。土壤中的有机物很大一部分来源于植物凋落物和根系分泌物,这为土壤中的微生物提供了丰富的养分,山地的植被生物量较大,对 SOC有较大影响(杨玉盛等,1998;秦杰等,2015)。在4种不同生境土壤中,细菌的丰富度和多样性指数均高于古菌的指数,表明古菌生态位的限制多于细菌,古菌对环境因子的利用限制性更高,Aller et al.(2010)也比较多种环境中细菌与古菌多样性的均表现为细菌的多样性高于古菌,本实验的研究结果与其一致。
原核生物群落的生态位分化主要体现在门水平上,何馨竹等(2021)在环青海湖农牧交错区的4种不同生境土壤细菌群落中,Proteobacteria、Actinobacteria、Acidobacteria、Gemmatimonadetes和Chloroflexi普遍存在于每一个生境的土壤中,它们也作为优势菌群出现在各类生境尤其是高寒土壤细菌群落(Hartmann et al.,2006;Hollister et al.,2010;Will et al.,2010;孙怀博,2013;候晓翠,2014;杨媛媛,2019),表明这些菌种可以很好地适应不同生境。不同生境间土壤细菌的群落组成结构存在显著差异,在农田(QSo)与山地(XSo)中 Proteobacteria的丰度最高且与其他生境相比到达显著水平,Proteobacteria作为嗜营养型菌(Singh et al.,2010),更易于在农田和冻土等植物凋落物积累的土壤中占据更广泛的生态位。Bacteroidetes作为农田(QSo)土壤中的独有优势种也可能受到土壤养分和农田作物根系分泌物的影响,而Actinobacteria在农田(DSo)和牧场(LSo)两种草地和牧场生境中占有最高的群落丰度,Actinobacteria作为富营养型细菌的代表,对土壤营养条件要求高并且对环境响应敏感,已有研究表明有效磷和碱解氮对 Actinobacteria的数量、种类分布都有显著影响(赵卉琳,2008),该结果进一步表明在环青海湖农牧交错区牧场和草地的土壤养分较为充足。Gemmatimonadetes和Chloroflexi的相对丰度在农田(DSo)中最高而Nitrospirae在山地(XSo)和牧场(LSo)中相对丰度较高。前人的研究表明Gemmatimonadetes能很好地适应各类生态系统,对土壤酶活性有积极响应,且在水分含量较低的环境更具有优势(Fawaz,2013;冯慧琳等,2021),Chloroflexi由于营养方式和代谢途径的多样性能参与土壤中碳、氮、硫多种循环因而也普遍存在于各类土壤中(鲜文东等,2020),在环青海湖农牧交错区,本研究采集土壤样品中的TN和AN含量较高,促进了Nitrospirae的繁殖。土壤微生物群落对pH变化响应敏感,pH值作为重要因素之一显著影着响土壤微生物群落组成结构(Bartram et al.,2014;Feng et al.,2014)。本研究中土壤细菌群落中 Acidobacteria的相对丰度与土壤pH值呈显著相关,随着pH的降低Acidobacteria的丰度呈现显著增加趋势,该结果与前人研究结果一致(Jones et al.,2009;Shen et al.,2013)。在本实验中,不同生境之间土壤的pH值存在显著差异,但是Acidobacteria的丰度却没有显著性差异,可能是由于Acidobacteria的丰度除了受pH值得影响,还受到其他环境因子的共同作用,控制原核生物群落结构和组成的机理机制还需要进一步探究。本实验中,土壤细菌的群落结构与pH、SOC、TN、Olsen-P、AN、AK含量和碳氮比都具有显著相关关系。在 4种生境的土壤古菌群落中,Thaumarchaeota和 Euryarchaeota是古菌群落中的两大主要门类。在前人的研究中,高寒冻土区优势古菌中包含 Euryarchaeota和 Crenarchaeota,在高寒草甸和高寒沼泽区Crenarchaeota都广泛分布(吴尊凤等,2012;李月娇等,2015;党宁,2020)。土壤古菌群落受到生境中的多种环境因子影响,环境因子中的pH值、SOC、TN、Olsen-P、AN、AK和碳氮比都显著作用于土壤古菌的群落组成。在4种不同生境土壤中,土壤环境因子的差异是影响土壤原核生物的群落组成结构和功能上的差异主要控制因素,反过来,关键微生物群落的差异也可作为土壤化学性质和养分状况的生物学参考指标。
利用分子生态网络对 4种不同生境土壤中原核生物群落相互作用进行研究,与细菌分子生态网络相比,古菌网络的连接数和节点数都明显较少,表明在同一生境古菌的生态位限制更高,古菌网络更易因环境因子的变化影响网络的结构(Kitano,2004;Wang et al.,2016;汪峰等,2014)。网络的节点数和连接数表现了网络的规模和关系复杂程度。在4个不同生境的细菌网络中,农田(QSo)和山地(XSo)的连接数更高,表明土壤细菌物种间的联系更加复杂,网络稳定性更高;并且在这2个网络中,Proteobacteria的节点数和连接数较高,这表明 Proteobacteria作为嗜营养菌在养分较高的环境中种间交互作用增加,而在草地(DSo)与牧场(LSo)中放线菌门Actinobacteria占主导地位。在古菌网络中山地(XSo)的连接数高于其他三者,可能由于古菌在养分充足,缺少人为干扰的环境能更好地定殖并维持群落的结构。这表明土壤化学性质、养分含量差异能够影响土壤中原核生物群落结构和不同菌种间的相互作用,这与李冰等(2020)研究对于不同土地利用方式土壤中微生物群落结构与土壤性质的关系得到的结果具有一致性。网络中的正相关和负相关可以用来表征微生物之间的相关关系,微生物间生态位一致或存在共生关系呈现正相关,若存在竞争或捕食关系则呈负相关(Layeghifard et al.,2017)。本实验中,在农田(QSo)和山地(XSo)土壤中细菌间正相关关系占大部分,尤其在农田(QSo)中,仅有正相关,表明在这 2种生境中土壤细菌群落间的合作关系大于竞争关系;在古菌群落中,山地(XSo)的古菌群落的正相关关系较多,农田(QSo)中以负相关关系为主,而草地(DSo)和牧场(LSo)中正负关系相当,这表明在农田、草地和牧场3种生境土壤中古菌间资源竞争更加激烈。与之相似,Barberán et al.(2012)研究发现151个土壤样品构成的细菌分子网络中,同一个门的OTUs趋向于彼此高度连接,表明细菌的亲缘关系越近越趋于彼此连接发生相互作用。
4 结论
环青海湖农牧交错区不同生境之间土壤化学性质和养分状况存在显著差异,人为干扰会引起土壤矿化使得有机养分含量降低,同时影响土壤中速效养分的含量。而不同生境土壤中原核微生物的丰度和多样性受到 pH、SOC、TN、AN、AK、C/N和Olsen-P影响显著,其中,农田土壤中细菌群落的丰度和多样性显著最高,古菌群落多样性表现为在山地土壤中群落丰度和多样性显著较高。细菌和古菌的群落结构和优势菌门在不同生境中存在一定差异,主要表现在 QSo中的特有优势细菌菌门为Bacteroidetes,XSo和LSo中Nitrospirae也是其中的优势细菌类群,古菌群落表现为DSo和XSo的优势菌群包括两种,分别是 Euryarchaeota和Thaumarchaeota,而QSo和LSo的优势菌门仅有一种为Thaumarchaeota。不同生境中土壤原核微生物的种间相互作用关系存在差异,菌种间的物质信息交流频率不同,原核微生物网络中低丰度菌门同样具有重要作用。
4种土壤类型中细菌和古菌的菌群丰度和多样性都处于不饱和状态,说明在环青海湖农牧交错区的高寒生态土壤中存有大量未被认知的原核生物群落和物种资源。为了获得更为完善的原核生物群落信息,充分发掘新物种的潜力,在今后的研究中还需加大采样的数量、加大测序广度和深度。此外,冗余分析结果显示原核生物群落结构与 4种不同土壤类型成对应关系,表明环青海湖农牧交错区地上生物因素与非生物因素一起控制着原核生物的群落丰度、多样性和组成结构。综上,本研究揭示了环青海湖农牧交错区原核生物群落的丰度和多样性,初步阐明土壤类型—原核生物群落的相互作用、相互影响的关系,为农牧交错区土壤质量和可持续发展提供了理论依据。
猜你喜欢
土壤学报(2022年3期)2022-08-26
大自然探索(2022年5期)2022-07-11
知识就是力量(2022年6期)2022-06-16
中国动物保健(2022年2期)2022-05-05
落叶果树(2021年6期)2021-02-12
北京大学学报(自然科学版)(2020年3期)2020-06-07
现代园艺(2018年1期)2018-03-15
体育科技(2016年2期)2016-02-28
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
安徽医科大学学报(2015年9期)2015-12-16
