
经典型Kaposi肉瘤合并获得性反应性穿通性胶原病一例
2021-10-11 06:50:28朱建建陈霄霄
实用皮肤病学杂志 2021年4期
朱建建,李 昕,陈霄霄,袁 佳,龙 剑,何 平,杜 维
Kaposi肉瘤(Kaposi sarcoma,KS)是一种少见的中间型血管内皮瘤,由奥地利皮肤病学家Kaposi于1872年首先报道,病灶多位于下肢,称之为特发性皮肤多灶性肉瘤[1]。该病在我国较为罕见,主要发生于新疆地区。而反应性穿通性胶原病(reactive perforating collagenosis,RPC)表现为变性的胶原从真皮穿出表皮的穿通性皮肤病,分遗传型和获得型。遗传型多累及儿童,获得性反应性穿通性胶原病(acquired reactive perforating collagenosis,ARPC)常发生于成人,多合并系统疾病[2],上述两种疾病同时发生于1例患者的报道较为少见,现报告如下。
临床资料
患者,男,56岁,湖南常德桃源县人。因全身红斑、丘疹伴瘙痒1年余,外阴紫红斑8个月,加重伴肿胀2个月,于2019年1月18日就诊。1年余前,无明显诱因患者躯干、四肢出现散在的绿豆至黄豆大小红斑及丘疹,伴瘙痒,无畏寒、发热等,按湿疹外用药物(具体不详)治疗后,瘙痒稍缓解,红斑、丘疹部分消退。8个月前,阴囊出现瘙痒性紫红斑,自行外用多种药物治疗后瘙痒稍缓解。5个月前外用药后出现阴囊糜烂、渗液,瘙痒加重并出现疼痛。门诊按湿疹样皮炎、接触性皮炎伴感染,给予甲泼尼龙注射液40 mg每日1次静脉滴注,枸地氯雷他定8.8 mg每日1次、多西环素胶囊100 mg每日2次口服治疗。1周后,阴囊肿胀、糜烂渗液明显减轻,干涸结痂,躯干红斑、丘疹部分消退,瘙痒减轻,糖皮质激素减量至醋酸泼尼松30 mg/d口服,患者拒绝进一步检查以明确诊断。醋酸泼尼松每5日减10 mg至停药,患者躯干部丘疹有所消退。2个月前,患者因上呼吸道感染后,躯干部丘疹复发增多伴瘙痒,外阴及四肢出现紫红斑、肿胀伴疼痛,出现排尿困难、尿少、尿中断等症状。患者自起病以来,无畏寒发热,精神食纳尚可,大便正常,体重无明显变化。既往有2型糖尿病、慢性乙型肝炎病史10余年,长期口服二甲双胍、阿卡波糖、格列齐特等药物降糖治疗,血糖控制基本稳定。否认疫区居住史;无输血、吸毒及冶游史。无药物过敏史,无毒物及有害物质长期接触史。系统查体:生命体征平稳,全身浅表淋巴结未触及增大,心、肺、腹无异常,双下肢轻度非凹陷性肿胀。皮肤科情况:躯干部及四肢散在红色丘疹及少量抓痕;阴囊及阴茎肿胀明显,阴囊紫红斑,表面有数个黄绿色圆形痂皮;左下肢胫前近膝关节处可见数个群集分布的紫红色孤立性结节,触之较硬,右下肢胫前散在分布紫色斑及丘疹;双手背、双足背非凹陷性水肿,双手第5指近端背侧、左足背可见边界清楚的紫红斑,皮温不高,压之不褪色(图1)。

图1 经典型Kaposi肉瘤合并获得性反应性穿通性胶原病患者临床表现
实验室及辅助检查:血常规中血红蛋白111.0 g/L(正常值130~175 g/L),嗜酸粒细胞百分率6.3%(0.4%~8%),动态红细胞沉降率83 mm/1 h(≤15 mm/1 h);血生化示白蛋白35.0 g/L(40~55 g/L),血 钾3.35 mmol/L(3.5~5.3 mmol/L),尿 酸500 μmol/L(180~440 μmol/L),乙型肝炎病毒(HBV)-DNA 1.14×104copy/ml(<100 copy/ml);肿瘤标志物铁蛋白407.19 ng/ml(0~322 ng/ml)。二便常规、肝肾功能、心肌酶、免疫相关检测、血糖、C反应蛋白、类风湿因子、抗O抗体、凝血功能均正常。丙肝病毒抗体、梅毒螺旋体血清学检测、人免疫缺陷病毒抗体(HIV-Ab)检测均正常。心电图示不完全性右束支传导阻滞。肺部、盆腔及阴囊计算机断层扫描示:双下肺部炎症,胸膜肥厚,纵隔及双侧腋窝淋巴结增大,双侧腹股沟淋巴结增大。腹部及双肾彩色超声多谱勒示:脂肪肝、脾稍大、左肾错构瘤、前列腺炎、双侧附睾头囊肿、右侧睾丸多发囊肿。双下肢动脉及深静脉彩色超声多谱勒未见明显异常。
阴囊及左小腿紫红斑组织病理示:真皮内肿瘤细胞呈团块状浸润,瘤体主要由梭形细胞和血管组成,梭形细胞纵横交错排列,有异形性,偶见核分裂像,瘤体内可见裂隙状管腔,腔内可见红细胞;梭形细胞间可见外渗的红细胞及含铁血黄素沉积,间质内少量炎性细胞浸润(图2)。免疫组化染色示:肿瘤细胞CD31、CD34、HHV-8阳性表达,D2-40部分表达,caldesmon阴性;核增殖相关抗原(Ki-67)约10%阳性(图3)。
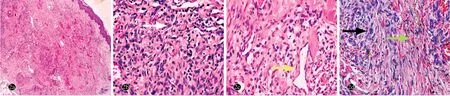
图2 经典型Kaposi肉瘤合并获得性反应性穿通性胶原病患者阴囊皮损组织病理
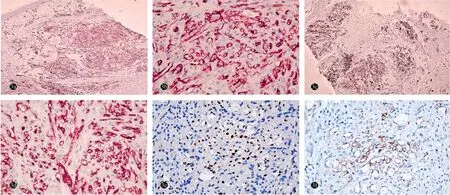
图3 经典型Kaposi肉瘤合并获得性反应性穿通性胶原病患者阴囊皮损免疫组化
背部丘疹组织病理示:表皮中央凹陷,内含角质物、淋巴细胞及变性的胶原,两侧可见角化不全及棘层稍增厚,其下方真皮内可见变性的胶原穿向表皮(图4a),Masson染色示蓝色胶原纤维穿出表皮(图4b),弹性纤维染色阴性。
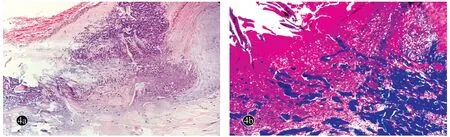
图4 经典型Kaposi肉瘤合并获得性反应性穿通性胶原病患者背部皮损组织病理
诊断:①经典型Kaposi肉瘤(阴囊、四肢紫红斑);②获得性反应性穿通性胶原病(背部丘疹)。治疗:甲泼尼龙注射液40 mg/d静脉滴注,雷公藤多苷20 mg每日3次、沙利度胺片20 mg每日2次、枸地氯雷他定8.8 mg每日1次口服,抗炎及止痒对症治疗。治疗2 d后躯干红斑有所消退,但瘙痒无改善。患者因经济原因回当地医院诊治(具体治疗方案不详),6个月后病情无好转,后失访。
讨论
Kaposi肉瘤(KS)临床上分4型:经典型或慢性地方型,获得性免疫缺陷综合征(AIDS)相关型或与AIDS相关的流行型,非洲地方型或淋巴结病相关型,医源性或移植相关型[3]。经典型多发生于>60岁的男性,病变主要位于下肢远端,尤其是足部[4]。该型患者主要集中在东欧和地中海沿岸,我国主要集中于新疆地区,病程平均为8~10年,发展较为缓慢,预后较好。本例为56岁男性,湖南常德人,无长期外地居住史,皮损发生于四肢及外阴,HIV-Ab为阴性,无移植及长期服用免疫抑制剂史,故属于经典型KS。
KS发病与HIV转换基因、细胞因子和人疱疹病毒8(human herpes virus 8,HHV-8)有关,肿瘤染色基因M和扩散因子在KS发病机制中有重要作用,常常与HIV转换基因产物和其他细胞因子[包括白细胞介素(IL)-1、肿瘤坏死因子(TNF)、IL-6、6FGF]共同作用[5]。尤其是HHV-8可能是KS重要的致病因子。张德志[6]研究表明KS 的高发病率与HHV-8的高感染率有一定的相关性;Henandez-Sierra等[7]指出,对于HHV-8血清学阳性受者,其KS发病风险比未受感染者高50倍。本例患者皮损免疫组化染色示HHV-8阳性表达,与大多数文献一致,证实了上述论断。
KS是多中心肿瘤,皮肤和黏膜常有多发性血管性结节,可从局限于四肢的皮肤表现发展至广泛的皮肤和内脏疾病。经典型病变主要位于下肢远端,尤其是足部,进展缓慢,可向肢体近端延伸。早期表现为淡红、紫罗兰色或蓝黑色斑或斑片,并融合成斑块状或结节状,表面可形成溃疡,常伴有肢体的非凹陷性水肿[4],但累及外生殖的报道少见。Attwa等[8]报道6例经典型KS患者中2例累及阴茎及阴囊,主要表现为红紫色到蓝色结节。其他类型如丘疹、斑块和疣状损害并不常见,常累及的部位是阴茎龟头、包皮、冠状沟、阴茎体、系带和尿道口,一些患者因积累面积较大而导致阴茎体积增大和淋巴水肿[9]。本例患者主要累及外生殖器,包括阴茎、尿道、包皮及阴囊,呈紫红斑及结节样改变,同时四肢及手足均出现淋巴水肿。
KS的组织病理主要表现为梭形细胞增生,纵横交错排列,裂隙样腔隙结构,红细胞外渗,含铁血黄素沉积以及慢性炎性细胞浸润。肿瘤细胞血管内皮细胞标志物常有不同程度的阳性,其中CD34、CD31最为稳定,HHV-8抗体阳性也有助于KS的诊断。根据其发展可分成3个可相互重叠的阶段:斑片期、斑块期及结节期。斑片及斑块期梭形细胞增生不明显,形态学缺少特异性,往往难以明确诊断。结节期病变可累及真皮全层甚至皮下组织,血管增生更明显,血管腔扩张,边缘不整齐,血管周围见增生的梭形细胞,梭形细胞可有一定异形性,梭形细胞之间红细胞外渗及含铁血黄素沉积较明显,炎性细胞浸润更密集[10]。本例患者皮损既有斑片、斑块也有结节表现,取材部位为结节处,具有结节期典型的组织病理改变,结合免疫组化染色均支持KS诊断。患者在本次就诊前曾出现过阴囊糜烂、渗出,确诊后分析,当时的症状可能就是KS不同阶段的临床表现,而并非单纯的刺激性接触性皮炎。前期四肢表现为淡褐或紫褐色斑片亦有可能是KS斑片期改变,但患者瘙痒症状明显,接诊者很容易误认为搔抓后导致的瘀斑或色素沉着。因此,在疾病早期阶段和对KS认识不足等一系列复杂情况下,KS极易被漏诊或误诊。
本例患者同时合并获得性反应性穿通性胶原病(ARCP)。ARCP是一种胶原经表皮排出的穿通性皮肤病,伴或不伴系统性疾病,临床表现为中心出现脐凹的丘疹,覆以不易剥脱的角栓及痂皮,全身瘙痒严重。组织病理学可见表皮缺损,痂皮内可见变性的胶原纤维由真皮乳头穿出表皮;Masson三色染色显示蓝色胶原纤维从由真皮穿出表皮,而弹性纤维染色为阴性[2]。成人期发病的患者多合并有严重的糖尿病、慢性肾衰竭、肝病、结核样麻风、艾滋病、霍奇金病、恶性肿瘤、肝炎、肝硬化、肾衰竭、肺纤维化、甲状腺功能减退或甲状旁腺功能亢进等[11]。本例患者有糖尿病、慢性乙型肝炎病史多年,可能与ARCP的发生有关,但与KS发病的关系尚不清楚。患者瘙痒及搔抓后继发皮损改变或KS早期的不典型等因素影响,无法区分二者发病的先后顺序,迄今为止尚无KS合并ARCP的报道。
KS需要与假性KS、梭形细胞血管瘤、Kaposi样血管内皮瘤、上皮样血管内皮瘤、血管肉瘤、黑素瘤等相鉴别,可通过组织病理、肿瘤细胞表达标志物、血管造影等方法进行区分。本例患者血管超声未见异常,组织病理见异形梭形细胞及裂隙样结构、HHV-8阳性这些特点可以与假性KS鉴别。
KS的治疗应依据不同临床类型制定个体化方案,但缺乏标准化指南,治疗远期疗效欠佳。其预后与临床类型、机体的免疫功能、疾病所处的阶段及有无机会性感染有关。经典型KS病程长,呈惰性发展,约2%播散至内脏[12]。本例患者从6个月后随访情况来看,KS病情进展较缓慢,ARCP在对症支持治疗后有所好转,但仍反复发作,可能与患者合并有慢性系统疾病相关。
猜你喜欢
高中数理化(2024年1期)2024-03-02 17:52:40
中国药学药品知识仓库(2022年8期)2022-05-09 13:54:24
基层中医药(2020年8期)2020-11-16 00:55:16
中学数学杂志(2019年1期)2019-04-03 00:35:42
广东教育·高中(2017年10期)2017-11-07 10:14:24
临床超声医学杂志(2017年5期)2017-06-19 19:22:29
西安建筑科技大学学报(自然科学版)(2016年5期)2016-11-10 02:39:26
中国卫生标准管理(2015年13期)2016-01-15 02:58:28
实用手外科杂志(2015年3期)2015-08-27 01:53:06
中国医疗美容(2015年2期)2015-07-19 10:11:59
