
运动诱导miRNAs调控PI3K/Akt/mTOR通路防治病理性心肌肥大的研究进展
2021-10-11 01:11田雪文王清路
中国体育科技 2021年9期
孟 昶 ,朱 磊 ,田雪文 ,王清路
2019年美国心脏协会报道,目前心血管疾病导致的死亡人数超过了所有癌症和慢性下呼吸道疾病的总和,是危害人类健康的主要因素(Targher et al.,2020)。同年我国国家心血管病中心数据显示,我国心脏病现患人数为2.9亿,死亡率居首位,占居民疾病死亡构成的40%以上,而病理性心肌肥大是一种很常见的心血管疾病,也是年轻人心脏骤停甚至猝死的常见原因(李惠钰,2019)。心肌肥大主要分为病理性和生理性,病理性心肌肥大通常与心功能障碍相关;生理性心肌肥大(运动诱导)不但不会影响心脏的正常功能,而且会增强心脏功能,保护心脏免受心脏病和心力衰竭的影响,被称之为“运动员心脏”。目前,关于防治心肌肥大较为成熟的通路有PI3K/Akt/Mtor(Ba et al.,2019)、IGF1-PI3K-Akt(Weeks et al.,2017)、Wnt/β-catenin(Zhang et al.,2016)、AMPK/FoxO/NFATc3(Samanta et al.,2020)以及 CaN/NFAT(Khalilimeybodi et al.,2018)等,其中PI3K/Akt/mTOR是经典信号通路。有研究表明,miRNAs可以调控机体内细胞代谢或细胞自噬(Unal et al.,2020),激活PI3K/Akt/mTOR通路(Ramasamy et al.,2015),达到防治病理性心肌肥大的效果(Wu et al.,2019)。近年研究发现,运动干预对miR-NAs调控PI3K/Akt/mTOR通路具有显著性影响(Ramasamy et al.,2015),但是具体的机制尚不明确。本研究通过综述运动、miRNAs、PI3K/Akt/mTOR通路及病理性心肌肥大四者之间的关系,探讨运动诱导miRNAs激活PI3K/Akt/mTOR通路的相关机制。
1 miRNAs概述
miRNAs是指长度约为22个核苷酸的内源性非编码单链RNA,通过与mRNA的3′-UTR区域结合,抑制mRNA的翻译,促进mRNA的降解,其独特的特点:一个miRNA可以同时调控几个基因,一个基因也可以同时受几个miRNAs调控(王世强等,2017)。现阶段研究发现,miR-21(Bai et al.,2015)、miR-144(Wen et al.,2020)和 miR-145(Zhu et al.,2018)等miRNAs可以通过激活PI3K/Akt/mTOR信号通路来防治病理性心肌肥大。
2 PI3K/Akt/mTOR通路
PI3K/Akt/mTOR通路是有关心肌肥大的经典信号通路(Cui et al.,2016),主要由PI3K、Akt和mTOR的3个作用分子组成,在正常细胞的增殖与凋亡中都发挥重要作用(Very et al.,2018)。PI3Ks是一组脂质激酶,在信号传导、细胞代谢和生存中起着关键性的作用,PI3K根据其结构特点和底物特异性可分为Ⅰ、Ⅱ、Ⅲ3种类型(Li et al.,2017b)。目前研究最广泛的是能被细胞表面受体激活的Ⅰ型PI3K。Ⅰ型又分为ⅠA和ⅠB两个亚型,ⅠA是催化亚基p110和调节亚基p85组成的异二聚体,PI3K Ⅰ A亚型由酪氨酸激酶受体、G蛋白偶联受体或其他炎症基因(如RAS)激活(Zhang et al.,2014)。活化后的PI3K可使细胞膜上的PIP2磷酸化为PIP3,PIP3去磷酸化为PIP2,该过程受抑制基因PTEN的负性调节,PIP3为信号蛋白磷酸肌醇依赖性蛋白激酶1(phosphoinositide dependent kinase-1,PDK1)和丝氨酸/苏氨酸激酶Akt提供锚定位点,一旦蛋白的THr308位点和Ser473位点分别被PDK1和PDK2磷酸化,则Akt被完全激活(Reddy et al.,2020)。激活后的Akt可启动信号通路下游的级联反应,如促进细胞生长、增殖、存活、迁移,以及细胞增殖等(Zhang et al.,2020a)。Akt可激活mTOR,导致新陈代谢改变、基因转录和翻译改变。mTOR是一种大的丝氨酸/苏氨酸蛋白激酶,属于磷脂酰肌醇激酶相关激酶家族,在进化中高度保守,但mTOR本身不具有酯激酶活性,而具有Ser/Thr蛋白激酶活性,能磷酸化蛋白底物的Ser/Thr残基,存在mTORC1和mTORC2两 种 不 同 的 复 合 体(Ortega et al.,2018)。mTORC1是Akt下游的靶点之一,Akt通过磷酸化脯氨酸富集区Akt基质1和结节性硬化症复合物2(tuberous scle-rosis complex 2,TSC2)激活mTORC1通过调控S6激酶调节蛋白质的合成,mTORC2则参与Akt Ser473位点的磷酸化过程(Wei et al.,2017)(图1)。

图1 PI3K/Akt/mTOR信号通路Figure 1.PI3K/Akt/mTOR Signaling Pathway
3 miRNAs调控PI3K/Akt/mTOR通路
miRNAs在调节心肌肥大过程中具有调节细胞凋亡的关键作用,特别是在调节心肌肥大细胞增殖和细胞死亡中效果显著。研究发现,miR-320(Wen et al.,2018)、miR-206(Chen et al.,2016)、miR-139(Zhang et al.,2020b)、miR-124(Zhu et al.,2019)、miR-21(Huang et al.,2017)、miR-144(Wen et al.,2020)、miR-145(Liu et al.,2017b)、miR-181b-5p(Chang et al.,2018)、miR-214(Liu et al.,2017a)、miR-93(Li et al.,2017a)以及 miR-496(Ji et al.,2020)与PI3K/Akt/mTOR通路显著相关。
有研究表明,miRNAs可以通过激活PI3K/Akt/mTOR通路来防治心血管疾病(Ramasamy et al.,2015;Samidurai et al.,2018;Zhang et al.,2017)。Yang等(2018)在心肌缺血再灌注损伤模型中,发现患有心肌缺血的H9C2细胞可以抑制miR-320的表达,并通过维持存活蛋白的表达来模拟胰岛素的心脏保护作用,从而激活PI3K/Akt/mTOR途径来对抗心肌缺血再灌注损伤。Kong等(2019)在缺氧条件下培养H9c2心肌细胞,建立心肌缺血再灌注损伤大鼠模型,探索核糖核酸内切酶失调对缺氧诱导的H9c2细胞损伤的影响。结果表明,核糖核酸内切酶的过表达加剧了缺氧诱导的H9c2细胞损伤,miR-206受到核糖核酸内切酶的负调控,而核糖核酸内切酶的过表达则通过miR-206的下调加重了缺氧损伤,研究还发现,ATG3是miR-206的靶基因,miR-206对缺氧损伤的作用是通过靶向ATG3来实现的。此外,在缺氧处理的H9c2细胞中,抑制核糖核酸内切酶的表达激活了PI3K/Akt/mTOR通路,而miR-206的过表达会逆转这一过程导致激活PI3K/Akt/mTOR通路失败,说明抑制核糖核酸内切酶可改善心肌功能,并抑制心肌缺血再灌注损伤后的细胞凋亡,即核糖核酸内切酶上调可能会加重心肌缺血再灌注损伤,也许是由于下调miR-206导致ATG3过表达实现的,PI3K/Akt/mTOR途径的激活可能是介导核糖核酸内切酶/miR-206/ATG3对心肌缺血再灌注损伤心脏保护的关键机制。Yu-an等(2020)研究了长链非编码RNA H19保护H9c2心肌细胞抵抗缺氧诱导的损伤,发现低氧的诱导使细胞活力降低,细胞发生迁移和侵袭,细胞凋亡增加以及H19的上调。敲除H19会增加低氧诱导的H9c2细胞损伤,并发现是通过上调miR-139加剧了缺氧引起的损伤。Sox8被确定为miR-139的靶基因,其表达受miR-139的负调控,上调Sox8的表达可能会激活PI3K/Akt/mTOR通路减少缺氧诱导的细胞损伤,说明增加长链非编码RNAH19的表达量会使miR-139的表达减少,导致Sox8的表达上升从而激活PI3K/Akt/mTOR通路减少缺氧诱导的细胞损伤。Ma等(2013)将Wistar雌性大鼠分为对照组和游泳锻炼组,游泳锻炼组的大鼠要每天完成1 h身体超负荷5%的游泳运动,每周5次,共8周,干预后发现与对照组相比,游泳锻炼组的心脏磷酸ser473-AKT和磷酸Ser2448-mTOR分别增加了46%和38%。游泳锻炼组miR-124降低了38%。在游泳锻炼组中,PIK3a(由miR-124靶向)的基因表达增加了213%,表明miR-124的减少可上调PIK3a基因从而诱导了PI3K/Akt/mTOR信号通路的激活。以上研究表明,抑制miR-320、miR-206、miR-139以及miR-124的表达可以激活PI3K/Akt/mTOR通路。
Ma等(2013)通过运动组与对照组相比发现,miR-21、miR-144和miR-145分别上调了152%、128%和101%。在运动组中PTEN蛋白(由miR-21和144靶向)和TSC2(被miR-145靶向)蛋白分别降低了51%和55%。说明miR-21和miR-144的表达增加会抑制PTEN水平,而miR-145的增加则可能抑制TSC2表达,从而诱导了PI3K/Akt/mTOR信号通路的激活。Chang等(2018)研究了miR-181b-5p在饥饿诱导心肌细胞自噬中的作用,发现下调miR-181b-5p可以促进心肌细胞自噬,另外miR-181b-5p还可以负调节Beclin-1和Hspa5基因的表达,而上调miR-181b-5p会通过Hspa5基因抑制自噬并促进细胞凋亡,萤光素酶报告基因测定的结果还证实Hspa5是miR-181b-5p的靶基因,上调miR-181b-5p可以直接抑制Hspa5通过PI3K/Akt/mTOR通路促进饥饿诱导的心肌细胞自噬和细胞凋亡。Chong等(2019)通过抑制miR-214观察缺氧条件下细胞的凋亡和自噬,发现东凌草素也是通过调节H9c2细胞中的相关蛋白来显著减轻缺氧诱导的细胞凋亡和自噬。在缺氧和东凌草素共同控制的细胞中发现了miR-214表达的上调,同时发现,miR-214的抑制作用消除了东凌草素在缺氧引起的细胞凋亡和自噬中的调节功能。此外,还发现PTEN是miR-214的靶基因,观察到东凌草素在低氧处理的H9c2细胞中通过miR-214升高激活PI3K/Akt/mTOR通路,说明东凌草素通过增强H9c2细胞中的miR-214表达,激活PI3K/Akt/mTOR通路来缓解缺氧引起的心肌细胞凋亡。Li等(2018)在血管紧张素II处理的心肌细胞中,发现心肌梗死相关转录物上调,miR-93下调。心肌梗死相关转录物充当心肌细胞中miR-93的分子筛,TLR4被确定为miR-93的靶基因,并且心肌梗死相关转录物通过使miR-93变分子筛促进了TLR4表达,TLR4的强制表达部分逆转了miR-93过表达对血管紧张素II诱导的心肌肥大的保护作用。此外,在血管紧张素II诱导的心肌肥大中,使心肌梗死相关转录物表达下降,会导致miR-93过表达,从而通过TLR4使PI3K/Akt/mTOR通路失活,说明抑制心肌梗死相关转录物可上调miR-93,导致TLR4基因被下调,从而抑制血管紧张素II诱导的心肌肥大。Jin等(2020)用过低氧复氧模型分析由急性心肌缺血引发的心肌梗塞,发现低氧复氧模型治疗H9c2细胞和心肌细胞凋亡显著减少都与miR-496的上调有关,双荧光素酶报告系统证实miR-496是钩微管束缚蛋白3的抑制因子,过表达的钩微管束缚蛋白3可刺激低氧复氧状态下处理细胞的凋亡从而降低细胞增殖,miR-496的上调可以激活PI3K/Akt/mTOR信号通路,在钩微管束缚蛋白3的帮助下,miR-496上调可保护细胞免受低氧复氧诱导的凋亡并刺激细胞增殖,miR-496靶向钩微管束缚蛋白3,激活PI3K/Akt/mTOR信号通路具有抗凋亡和增殖作用。以上研究表明,促进miR-21、miR-144、miR-145、miR-181b-5p、miR-214、miR-93 以 及miR-496的表达可以激活PI3K/Akt/mTOR通路。
通过以上研究发现,miRNAs可以激活PI3K/Akt/mTOR通路。抑制miR-320、miR-206、miR-139以及miR-124的表达可以促进或维持其靶基因的表达,从而抑制细胞损伤或细胞凋亡达到激活PI3K/Akt/mTOR通路的效果;促 进 miR-21、miR-144、miR-145、miR-181b-5p、miR-214、miR-93以及miR-496的表达可以促进或抑制其靶基因的表达,从而促进线粒体自噬或细胞增殖以及抑制细胞凋亡,从而达到激活PI3K/Akt/mTOR通路的效果(图2)。这些miRNAs和PI3K/Akt/mTOR通路很有可能成为病理性心肌肥大与心血管疾病的潜在治疗靶点,对心血管疾病的防治具有不可忽视的作用。

图2 miRNAs对PI3K/Akt/mTOR通路的调控Figure 2. Regulation of PI3K/Akt/mTOR Pathway by miRNAs
4 运动诱导miRNAs
运动可通过减少脂肪细胞数量来降低体质量,增加胰岛素敏感性以及葡萄糖摄取,肌肉力量和耐力,抗氧化剂水平和HDL,降低LDL和TG、TC(孟昶 等,2019;朱磊等,2019)。从心血管角度看,运动训练可以降低舒张压和收缩压,增加左心室射血分数,改善血管功能,增加心肌质量,这被称为运动训练引起的心肌肥大(Fernandes et al.,2015;Weeks et al.,2017)。有氧运动可诱发有益的生理性左室重塑,目前,心脏研究已鉴定出与有氧运动相关的抗心肌肥大型 miRNAs,如 miR-1(Unal et al.,2020)、miR-133(Diniz et al.,2015)、miR-26(Zhang et al.,2013)、miR-9(Wang et al.,2010)、miR-98(Yang et al.,2011)、miR-29(Shieh et al.,2011)、miR-378(Yuan et al.,2018)和miR-145(Li et al.,2013)等,促心肌肥大型 miRNAs,如miR-143(Sun et al.,2019)、miR-103(Qi et al.,2019)、miR-130a、miR-146a(Chavali et al.,2014)、miR-21(Tomaniak et al.,2018)、miR-210(Hirt et al.,2015)、miR-221(Kakimoto et al.,2018)、miR-222(Hirt et al.,2015)、miR-27a/b(Her-nandez-Torres et al.,2014)、miR-199a/b(Li et al.,2016)、miR-208(Soci et al.,2016)、miR-195(You et al.,2014)、miR-499(Shieh et al.,2011)、miR-34a/b/c(Ooi et al.,2017)、miR-497(Qi et al.,2020)、miR-23a(Hernandez-Tor-res et al.,2014)和 miR-15a/b(Zhang et al.,2016)等。有研究表明,游泳运动可以调控miR-143、miR-144、miR-145、miR-208a和miR-222增加心肌细胞的生长和存活率,还可以调控 miR-1、miR-26、miR-27a、miR-133、miR-143、miR-150和miR-222影响心脏重构和血管生成等(Chavali et al.,201;Fernades et al.,2015;Hirt et al.,2015)。综上所述,miRNAs在心脏保护作用方面具有潜在作用,也足以证明运动可以诱导miRNAs的表达从而影响心血管疾病的发生发展。
5 运动调控PI3K/Akt/mTOR通路
PI3K/Akt/mTOR通路可以调节细胞生长、代谢、存活和血管生成,而运动可以调控此通路。Yin等(2020)研究表明,将48只7周龄的SD大鼠进行了3周中等强度有氧运动,可以选择性地提高IGF-1、IGF-1R和mTOR的水平以及PI3K和Akt的活性。Bao等(2020)调查了有氧耐力运动对老年小鼠肾脏血管硬化的保护作用及PI3K/Akt/mTOR通路的作用,发现有氧耐力运动能保持老年小鼠的肾脏形态和肾功能,肾小球基底膜厚度明显增加,足细胞足突消失,有氧耐力运动还显著改善了整个病变范围。在老年对照组中,血管内皮生长因子和JG12的蛋白表达较低;有氧耐力运动组中,血管内皮生长因子、JG12和Bcl-2蛋白表达显著增加,Bax、Caspase 3、IL-6和衰老细胞的表达降低,从而导致PI3K及其下游信号分子Akt和mTOR的上调,这一研究结果与Yin等(2020)一致。Kang等(2015)将实验动物分为非转基因对照组和转基因运动组(跑台训练12周,每周5次,每次1 h),干预后进行水下迷宫认知功能检测,发现转基因运动组Beclin-1蛋白表达增加,p62蛋白表达降低,PI3K/Akt/mTOR通路通过增加磷酸化得到改善(其活性受到GSK-3β磷酸化的抑制),这一研究结果同样证实了运动对PI3K/Akt/mTOR通路的调控作用。Ma等(2013)将雌性Wistar大鼠进行游泳训练,训练8周,每周5次,每次1 h,发现游泳运动增加了PIK3a和磷酸化Thr1462-TSC2蛋白表达,同时降低了PTEN和TSC2蛋白表达,从而诱导了PI3K/Akt/mTOR通路的激活,再一次证明了运动对PI3K/Akt/mTOR通路的调控作用。上述研究表明,运动可以通过改变靶向基因的表达激活PI3K/Akt/mTOR通路。
6 运动对病理性心肌肥大的防治作用
心脏由心肌细胞、非肌细胞(如成纤维细胞、内皮细胞、肥大细胞、血管平滑肌细胞)和周围的细胞外基质组成(Olah et al.,2019)。心室心肌细胞仅占心脏细胞总数的1/3,但占心脏质量的70%~80%,生理和病理刺激都会导致心脏体积的增大,这主要是由于心肌细胞体积的增大,病理性心肌肥大通常与心肌细胞丢失和纤维化替代、血管生成不足、心功能障碍、心力衰竭以及猝死相关;与之相反,生理性心肌肥大与正常的心脏结构、维持或增强的心脏功能相关,可以保护心脏免受心脏病和心力衰竭的影响。运动防治病理性心肌肥大是一种重要的非药物性方法,可以延长心肌细胞的寿命,增加心室搏动量和心输出量,从而改善有氧能力(Olah et al.,2019;Rahimi et al.,2018)。Luckey等(2017)对雌性病理性心肌肥大小鼠模型进行运动研究,发现运动诱导的心肌肥大(生理性肥大)与心脏功能正常或增强有关,没有明显的纤维化和细胞凋亡,还能同时诱导脂肪酸和葡萄糖氧化,刺激生长激素的释放,该过程会激活心脏中许多细胞受体和细胞内信号传导途径(如PI3K/Akt/mTOR信号传导途径)。Soares等(2019)将雄性小鼠分为对照组、不超负荷运动组(每天游泳2次)和超负荷运动组(每天游泳3次),共训练6周,虽然发现两个运动组都出现了心肌肥大状况,但未观察到纤维化,并提高了氧化能力,这一研究结果与Luckey等(2017)一致。Baghaiee等(2018)将20只wistar大鼠进行8周有氧运动后发现,8周的训练使小鼠血浆Klotho蛋白(抗衰老)水平显著升高,左心室内径显著增大,左心室壁厚和纤维化程度显著下降,表明适度的有氧训练可以通过恢复Klotho的水平,减轻氧化应激和减缓心肌衰老,缓解病理性心肌肥大,同样对运动可以防治心肌肥大提供了理论依据。Qi等(2020)对运动防治心肌肥大的机制进行了更深入的研究,发现运动诱发的生理性心肌肥大心脏中自噬活性显著增强,IGF-1可以刺激H9C2心肌细胞变肥大,miR-26b-5p、miR-204-5p和miR-497-3p过表达可以通过抑制自噬而显著减弱IGF-1诱导的H9C2细胞肥大,miR-26b-5p、miR-204-5p和miR-497-3p分别通过靶向ULK1、LC3B和Beclin 1减弱了H9C2细胞的自噬,表明运动可以通过调节miR-26b-5p、miR-204-5p和miR-497-3p减弱H9C2心肌细胞的自噬,从而诱导生理性心肌肥大。综上所述,适度的运动可以导致生理性心肌肥大,不会导致心脏功能的下降,还会提高氧化能力防止纤维化,最重要的是可以防治病理性心肌肥大。
7 运动诱导miRNAs调控PI3K/Akt/mTOR通路防治病理性心肌肥大
目前,国外已有关于运动诱导miRNAs调控PI3K/Akt/mTOR通路防治心血管疾病的相关研究,但是采用的手段和方法还较为单一,因此得出一个肯定的结论似乎还具有一定的困难。有研究发现,运动促进心脏健康、产生心肌保护效应与运动促进细胞增殖、抑制心肌细胞凋亡有关(Ramos et al.,2018),而PI3K/Akt/mTOR通路一般会出现激活或者抑制的变化趋势,PTEN和TSC2是PI3K/Akt/mTOR通路中的两个重要调节剂(Maehama et al.,1998),其调节机制与miRNAs的参与有关(Cheng et al.,2007)。
PTEN是PI3K/Akt/mTOR通路的主要负调节剂,它在许多细胞功能中发挥重要作用,如细胞迁移(Leslie et al.,2007),血小板生长因子诱导的膜皱褶(Leslie et al.,2000),胰岛素敏感性(Lackey et al.,2007),以及致癌作用等,据报道横纹肌特异的PTEN缺失,小鼠的心脏质量会增加约50%。TSC2类似于PTEN,它还是mTOR的抑制剂,是PI3K/Akt/mTOR信号转导的另一个负调节剂,Akt激酶使TSC2磷酸化会促进其与TSC1的解离,从而激活mTOR,导致蛋白质合成增加以及细胞大小增大(Inoki et al.,2002)。有研究显示,PTEN受miR-21和miR-144的调节,TSC2受miR-145的调节,Ma等(2013)研究表明,游泳锻炼组与对照组相比心脏中Akt和mTOR的表达显著增加,miR-21、miR-144和 miR-145均显著增加 ,PTEN 和TSC2的蛋白表达水平显著降低,但是PI3K/Akt/mTOR通路被激活,这表明8周的游泳运动有助于PI3K/Akt/mTOR通路的激活。另一项研究同样发现,与对照组相比,游泳锻炼组的生理性心肌肥大中PTEN蛋白水平降低,PTEN在调节大鼠心肌肥大中发挥了显著作用。因此,PTEN可能参与了游泳运动诱导的生理性心肌肥大。目前,在运动引起的心肌肥大中鲜见针对miRNAs调节PI3K/Akt/mTOR通路的研究,而miRNAs其特点是靶向多个基因,但靶向的基因受特定miRNAs的控制,有些miRNAs的表达减少表明靶基因的改善,miR-144似乎是这种情况。Palabiyik等(2019)将8只成年雄性SD大鼠进行每天1 h,每周5天,共4周的游泳运动干预,发现游泳锻炼组与对照组相比,PI3K、Akt、mTOR基因和蛋白质的表达以及miR-144的表达显著增加,而PTEN的基因和蛋白质的表达水平显著降低,说明游泳运动似乎会影响miR-144以及PI3K/Akt/mTOR通路。Liu等(2017b)进一步验证了此实验,发现游泳锻炼组和对照组相比,靶基因PTEN和TSC2的表达水平较低,而miR-144和miR-145表达增加,这一事实证明miR-144和miR-145的表达增加会抑制靶基因PTEN和TSC2的表达。Subbiah等(2015)对SD大鼠进行连续8周的游泳训练建造生理性心肌肥大模型,随后分离总RNA,并进行小RNA测序,对小RNA读数的分析揭示了生理性肥大过程中大量miRNA的差异表达,通过qPCR验证了显著差异表达的miRNA表达谱,再通过miRanda、miRdB和TargetScan在计算机模拟中预测靶基因,最后再用 qPCR 分析揭示了 miR-19b、miR-30、miR-133b、miR-208b、miR-22、miR-99b、miR-100、miR-181a和miR-191a的靶向基因,如miR-19b在生理性心肌肥大过程中显著上调,靶向基因是PTEN、MuRF、Bcl2、Atrogin-1和αCryB,通过靶向Atrogin-1和MuRF-1来积极调节心肌肥大,靶向Bcl2来增加细胞存活率并负面调节细胞凋亡;miR-30在生理性心肌肥大过程中显著上调,靶基因是CaMKIIδ、Eg-fr1和Bcl2,在病理性心肌肥大过程中被血管紧张素II诱导显著下调,激活钙信号传导细胞凋亡和自噬,进而通过过度自噬诱导病理性心肌肥大;miR-133b在生理性心肌肥大过程中显著上调,靶基因是CyclinD、Nelf-A、RhoA和Ccd42;miR-208b在生理性心肌肥大过程中显著上调,靶基因是THRAP1和Myostatin,可改善病理性心肌肥大引起的心力衰竭;miR-22在生理性心肌肥大过程中表达下调,靶基因是CDK6、Sir1和Sp1,miR-22可以诱导心肌肥大,是心肌肥大和重塑的关键调节剂,对调节细胞增殖、细胞分化和病理性心肌肥大起到关键作用;miR-99b和miR-100在生理性心肌肥大过程中被高度下调,其靶基因IGF1R、Akt和mTOR表达上调;miR-181a在生理性心肌肥大过程中表达下调,通过干扰NF-κB功能和细胞增殖来保护系统免受炎症损伤;miR-191a在生理性心肌肥大过程中表达下调,其靶基因mRNA-p53表达上调。上述差异表达miRNAs及其靶基因主要通过PI3K/Akt/mTOR和MAPK信号通路调控心肌细胞凋亡和死亡的结论。以上研究表明,运动可以影响miRNAs的表达,从而调节其特定的靶基因激活PI3K/Akt/mTOR通路,并在运动介导的生理性心肌肥大中起到关键作用,而运动诱导的生理性心肌肥大可以增强心脏功能,保护心脏免受心脏病和心力衰竭的影响,具有缓解病理性心肌肥大的效果,证明运动诱导miRNAs调控PI3K/Akt/mTOR通路防治病理性心肌肥大似乎是可行的。
综上所述,运动可以诱导miR-21、miR-144、miR-145、miR-19b、miR-30、miR-133b和 miR-208b表达上调,miR-22、miR-99b、miR-100、miR-181a和 miR-191a表达下调激活PI3K/Akt/mTOR通路防治病理性心肌肥大。目前多数参与运动防治病理性心肌肥大的miRNAs只停留在知道参与的层面上,具体的调控机制尚不明确,还需进一步探究PI3K/Akt/mTOR通路中涉及的miRNAs和靶基因与运动之间的关系。现阶段关于miR-21、miR-144和miR-145的研究较为完善,其靶基因PTEN和TSC2是PI3K/Akt/mTOR通路的主要负调节剂,PTEN受miR-21和miR-144的调节,TSC2受miR-145的调节,运动又可以提高miR-21、miR-144和miR-145的表达,因此得出:运动可以诱导miR-21、miR-144和miR-145表达的增加,使其靶基因PTEN和TSC2表达降低从而激活PI3K/Akt/mTOR通路防治病理性心肌肥大(图3)。
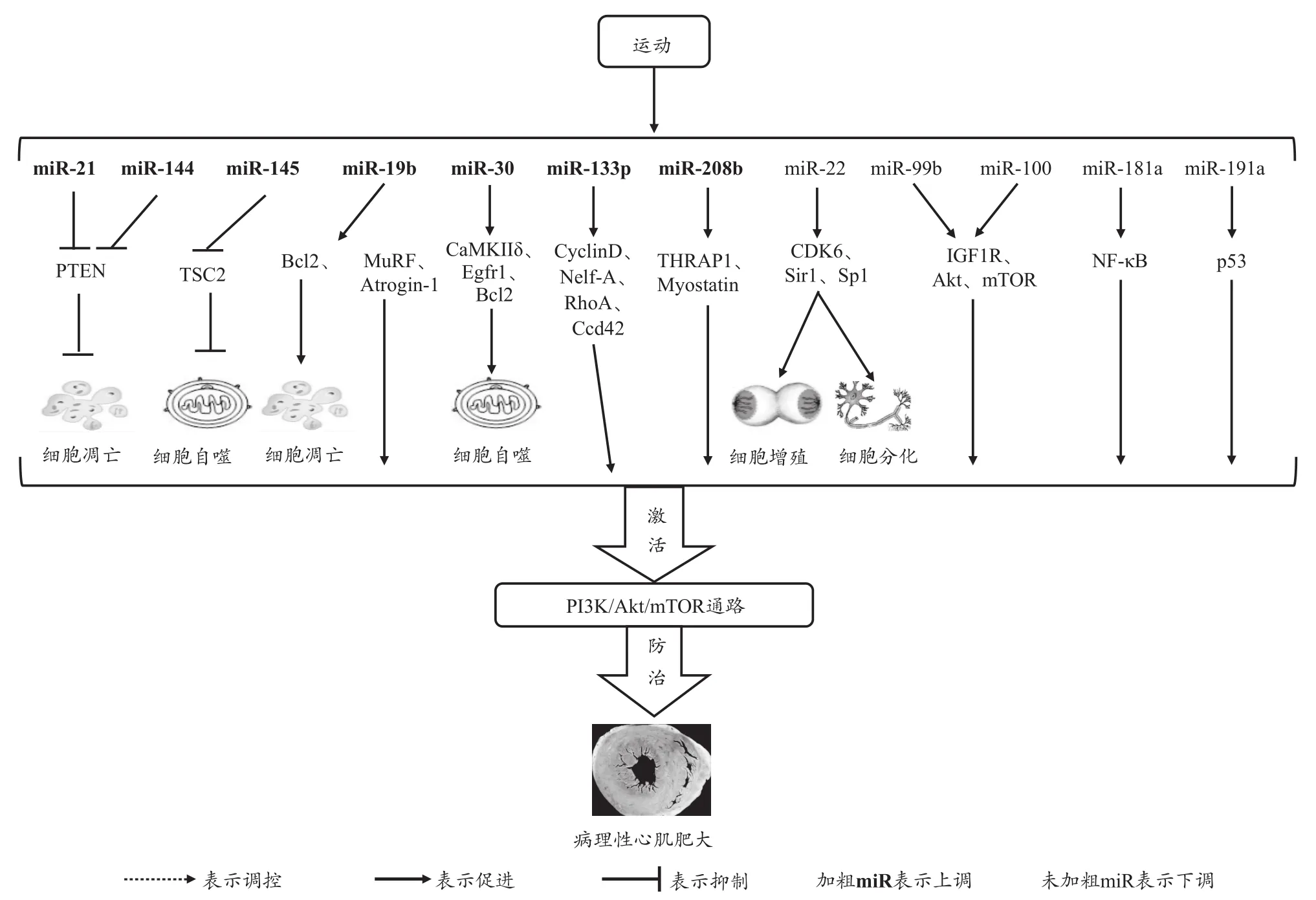
图3 运动诱导miRNAs激活PI3K/Akt/mTOR通路防治病理性心肌肥大机制Figure 3.Exercise-induced miRNAs Activate PI3K/Akt/mTOR Pathway to Prevent Pathological Myocardial Hypertrophy
8 结论与展望
近年,人们对运动的观念发生了转变,除了医药因素外,运动因素在心血管疾病防治中的作用逐渐强化。研究发现,运动可以诱导miR-21、miR-144和miR-145表达上调,使其靶基因PTEN和TSC2表达降低,激活PI3K/Akt/mTOR通路防治病理性心肌肥大。目前关于运动诱导miRNAs调控PI3K/Akt/mTOR通路对心血管疾病的防治主要聚焦于对心肌肥大影响的研究,但是具体的机制尚不完善,还需进一步探究PI3K/Akt/mTOR通路中涉及的其他miRNAs和靶基因与运动之间的关系。
猜你喜欢
中西医结合心脑血管病杂志(2022年18期)2022-10-21
中国畜牧兽医(2022年9期)2022-09-22
健康体检与管理(2022年4期)2022-05-13
牡丹江医学院学报(2022年1期)2022-01-06
新农民(2021年17期)2021-09-16
中国药学药品知识仓库(2021年18期)2021-02-28
新一代(2020年22期)2020-10-09
青岛大学学报(医学版)(2020年5期)2020-09-29
妇女(2020年4期)2020-04-20
知识窗·教师版(2019年7期)2019-09-26
