
基于三维荧光光谱及模式识别技术对当归质量的分析与鉴别
2021-10-09 05:19:48郭晴茹刘红石振萍杨扶德石晓峰邵士俊
现代食品科技 2021年9期
郭晴茹,刘红,石振萍,杨扶德,石晓峰,邵士俊*
(1.中国科学院兰州化学物理研究所,中国科学院西北特色植物资源化学重点实验室,甘肃省天然药物重点实验室,甘肃兰州 730000)(2.中国科学院大学存济医学院,北京 100093)(3.甘肃中医药大学药学院,甘肃兰州 730000)(4.甘肃省医学科学研究院,甘肃兰州 730050)
三维荧光光谱提供了荧光强度随激发波长和发射波长同时变化的荧光信息,也称为全扫描荧光光谱,它能够完整地描述物质的荧光特征,具有灵敏度高、选择性好等优点,且获得的三维荧光等高线图具有指纹性[1,2]。三维荧光光谱分析技术已在石油化工、医药、环保等领域得到广泛应用[3-5]。针对植物鉴别与质量评价,三维荧光指纹图谱具有能够反映多组分复杂体系整体化学特征的独特优势,极具应用潜力[6]。
当归为伞形科植物当归[Angelica sinensis(Oliv.) Diels]的干燥根,味甘、辛,性温,归肝、心、脾经,具有补血活血、调经止痛、润肠通便之功效[7],有“补血圣药”之称,在调节人体血液功能、保肝强肾健体等方面具有重要的药理作用。在我国传统饮食文化中,当归作为香辛料和调味品被广泛食用,美国、欧盟、日本也将当归作为香辛料食用。作为一种常用的大宗药食同源物质,当归的食品安全与质量控制至关重要[8-14]。当归的化学成分可分为苯酞类、香豆素类、有机酸类,以及氨基酸、多糖、维生素和微量元素等多种类别,目前,主要采用紫外光谱、荧光光谱、红外光谱、高效液相色谱等方法对当归的化学成分/组分进行定性定量分析,用于当归质量控制与评价,将三维荧光光谱分析挤模式识别技术应用于当归质量分析与鉴别的研究鲜有报道[15-19]。本工作结合当归所含化学成分,如苯酞类、黄酮类、生物碱类、氨基酸和蛋白质等所具有的光谱性质,研究建立当归水溶性、醇溶性和脂溶性提取物的三维荧光光谱分析方法,并将三维荧光光谱信息与计算机模式识别技术相结合,应用于当归质量一致性评价和真伪鉴别,为当归快速鉴别和质量评价提供参考。
1 材料与方法
1.1 仪器
Lambda 35 紫外可见分光光度计,PerkinElmer 公司;Fluorolog-3 荧光光谱仪,HORIBA 公司;PB-10/C标准型pH 计,Sartorius 公司;JB-040S 洁盟牌超声波清洗机,深圳市洁盟清洗设备有限公司。
1.2 材料
当归、欧当归样本分别采集于甘肃岷县、漳县、渭源、陇西、宕昌、天祝、临潭和青海湟源等种植基地(见表1),经甘肃中医药大学杨扶德教授鉴定为伞形科植物当归[Angelica sinensis(Oliv.) Diels]的干燥根、伞形科植物欧当归(Levisticum officinalisKoch)的干燥根。伞形科植物川穹、独活、北沙参、蛇床子和小茴香购买于兰州市药店,水为超纯水,其它试剂均为分析纯。
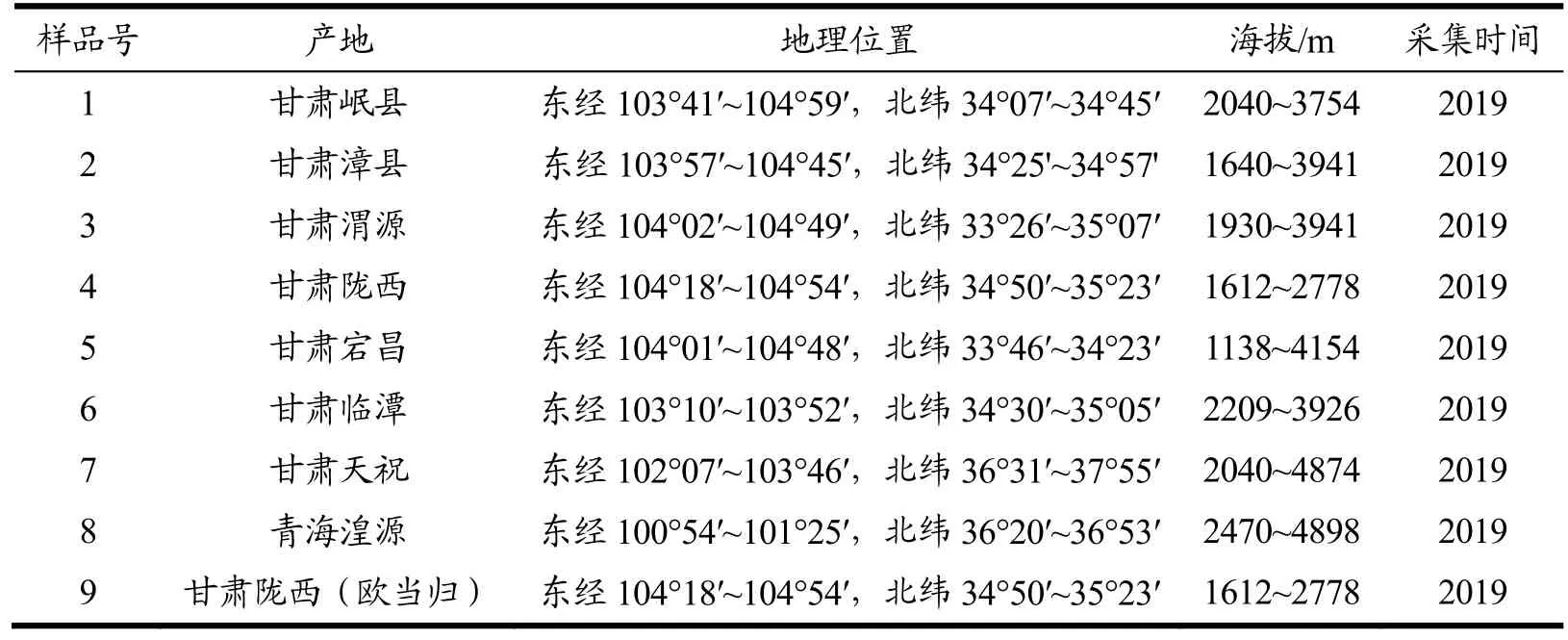
表1 不同产区当归样品 Table 1 Angelica sinensis samples from different producing areas
1.3 方法
以采自甘肃岷县的当归药材为样本,进行当归特征提取物制备条件和光谱分析条件的筛选优化实验,系统考察影响因素,建立供试品溶液制备及三维荧光光谱分析方法。
1.3.1 供试品溶液制备
1.3.1.1 水提取物
当归粉碎,过40 目筛,准确称取5.00 g,加入100 mL水,浸泡30 min后,油浴磁力搅拌回流提取30 min,冷却后,补齐重量,经离心分离,取上清液作为母液备用(浓度50.00 mg/mL,即每毫升相当于50.00 mg药材)。用超纯水稀释,配制成适当浓度的供试品溶液,用于光谱分析。
1.3.1.2 甲醇-水提取物
当归粉碎,过40 目筛,准确称取2.00 g 于具塞三角瓶中,加入甲醇-水(V:V,1:1)40 mL,超声提取45 min,离心分离,取上清液作母液备用(浓度50.00 mg/mL,即每毫升相当于50.00 mg 药材)。用50%甲醇-水溶剂稀释,配制成适当浓度的供试品溶液,用于光谱分析。
1.3.1.3 乙酸乙酯提取物
当归粉碎,过40 目筛,准确称取5.00 g 于索氏提取器中,加入100 mL 乙酸乙酯,加热回流2 h,抽提至抽取筒内溶剂无色,提取液转移至圆底瓶中,冷却后补齐重量,作为母液备用(浓度50.00 mg/mL,即每毫升相当于50.00 mg 药材)。用乙酸乙酯稀释,配制成适当浓度的供试品溶液,用于光谱分析。
1.3.2 缓冲溶液配制
称取磷酸氢二钠9.47 g 和磷酸二氢钾9.07 g 分别加入1000 mL 的容量瓶中,加蒸馏水溶解并定容,贮存在4℃的冰箱中,用时分别按不同比例混合,利用酸度计调控pH 值,配制pH 4.0~pH 9.0 一系列溶液。强酸(pH 2.0)或强碱pH(pH 11.0)分别用1.2 mol/L HCl 溶液或1.0 mol/L NaOH 溶液调配。
1.3.3 吸收和荧光光谱测试
室温下测定供试品溶液的紫外可见吸收光谱和荧光光谱。三维荧光光谱测试条件为:激发波长(λex)范围250~400 nm,采样间隔10 nm,激发狭缝2 nm;发射波长(λem)范围270 nm~600 nm,采样间隔10 nm,发射狭缝2 nm;积分时间0.2 s。
1.3.4 数据处理
为减小实验误差每个样品测定2~5 次,采用Origin 整理各次测定得到的三维荧光光谱图中相对应的数据,建立判别函数,采用SPSS 22 软件进行数据处理和判别分析,差异显著水平为p<0.05。
2 结果与讨论
2.1 当归提取物的三维荧光光谱
在优化的供试品制备方法和光谱测试条件下,当归水提取物、50%甲醇-水提取物和乙酸乙酯提取物的紫外吸收光谱和三维荧光光谱分析结果如图1 所示。
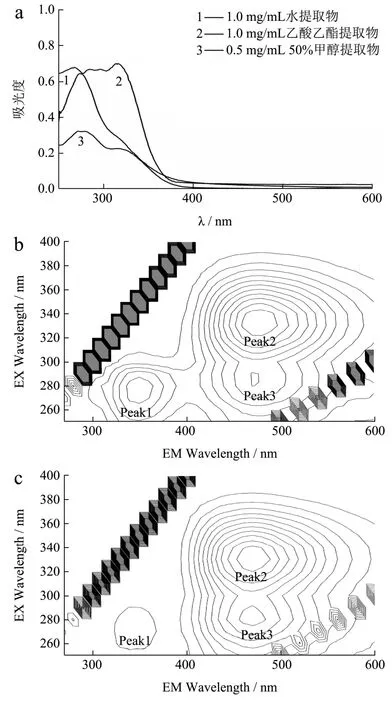

图1 当归提取物的紫外吸收光谱和三维荧光光谱 Fig.1 Absorption spectra and three-dimensional fluorescence spectra of Angelica sinensis extracts
当归水提取物(1.00 mg/mL)的最大吸收峰出现在270 nm,并有325 nm 的肩峰;三维荧光光谱呈现出3 个特征激发/发射(λex/λem)峰,即270 nm/350 nm(Peak 1)、330 nm/470 nm(Peak 2)和280 nm/470 nm(Peak 3)荧光峰。50%甲醇-水提取物(浓度0.50 mg/mL)的紫外吸收光谱和三维荧光光谱特征与水提取物类似,但吸收光谱的325 nm 吸收明显增强;三维荧光光谱的3 个特征激发/发射峰中,Peak 2 和Peak 3 的荧光强度相对于Peak 1 显著增强。结果表明,当归水提取物与50%甲醇-水提取物的荧光物质成分组成相近,但后者形成475 nm 发射峰(Peak 2 和Peak 3)的荧光物质成分含量较高。当归乙酸乙酯提取物具有完全不同于水提取物和甲醇-水提取物的吸收和发射光谱,三维荧光光谱呈现出2 个激发/发射特征峰,分别是265 nm/295 nm(Peak 4)和325 nm/425 nm(Peak 5)荧光峰,吸收和发射光谱的差异反映了脂溶性提取物具有不同的荧光物质成分。
2.2 提取条件优化
2.2.1 水提取物
考察提取溶剂水的用量(固液比分别为1:5、1:10、1:20、1:40),提取时间(15 min、30 min、45 min)对荧光光谱影响,结果如图2 所示。以浓度1.00 mg/mL供试品溶液三维荧光光谱特征峰的相对强度来判定,确定固液比1:20,提取时间30 min为最佳的提取条件。
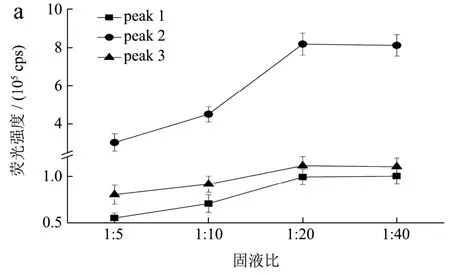
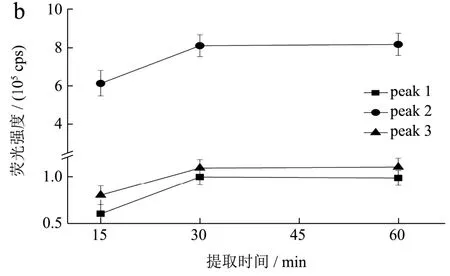
图2 不同溶剂用量和提取时间的当归水提取物三维荧光光谱特征峰强度变化 Fig.2 Fluorescence intensity changes of the three-dimensional fluorescence spectral characteristic peaks of Angelica sinensiswater extract with different solvent dosage and extraction time
2.2.2 甲醇-水提取物
考察提取溶剂甲醇-水的配比(0:1、1:1、1:0)、用量(固液比1:10、1:20、1:40)和提取时间(30 min、45 min、60 min)的影响,结果如图3 所示。以浓度0.50 mg/mL 供试品溶液三维荧光光谱特征峰的相对强度来判定,确定甲醇-水体积比1:1,固液比1:20,提取时间45 min 为最佳的提取条件。


图3 不同甲醇-水配比、溶剂用量和提取时间的当归提取物三维荧光光谱特征峰强度变化 Fig.3 Fluorescence intensity changes of the three-dimensional fluorescence spectral characteristic peaks of Angelica sinensis extracts with different methanol-water ratio,solvent dosage and extraction time
2.2.3 脂溶性提取物
考察提取溶剂(石油醚、正己烷、乙酸乙酯)和提取时间(1 h、2 h、3 h)的影响,结果如图4 所示。以浓度1.00 mg/mL 供试品溶液三维荧光光谱特征峰的相对强度来判定,确定乙酸乙酯抽提2 h 为最佳的提取条件。

图4 不同提取溶剂和提取时间的当归脂溶性提取物三维荧光光谱特征峰强度变化 Fig.4 Fluorescence intensity changes of the three-dimensional fluorescence spectral characteristic peaks of Angelica sinensisfat-soluble extracts with different extraction solvent and extraction time
2.3 供试品溶液浓度的影响
根据荧光分析的一般规律,稀溶液中荧光强度与荧光物质的浓度成正比,荧光物质浓度较大时会发生自猝灭,使荧光强度降低。将当归提取物的母液配制成不同浓度的供试品溶液,考察浓度对体系紫外吸收光谱和三维荧光光谱特征峰的影响。
不同浓度的水提取物紫外吸收光谱和三维荧光光谱特征峰荧光强度变化见图5。结果表明,在考察的浓度范围内(0.50~4.50 mg/mL),当归水提物紫外吸收强度随浓度升高而增强;三维荧光光谱中Peak l、Peak 3 特征峰的荧光强度受浓度影响较小,Peak 2 荧光强度随浓度升高而逐渐增强并趋于平稳,考虑到特征峰之间荧光强度的差异以及所得三维荧光光谱图应最大限度总体反映荧光物质信息,确定供试品溶液最佳检测浓度为1.00 mg/mL。50%甲醇提取物的紫外吸收与三维荧光光谱特征峰发射强度随供试品溶液浓度的变化与水提取物的类似,供试品溶液最佳检测浓度为0.50 mg/mL。
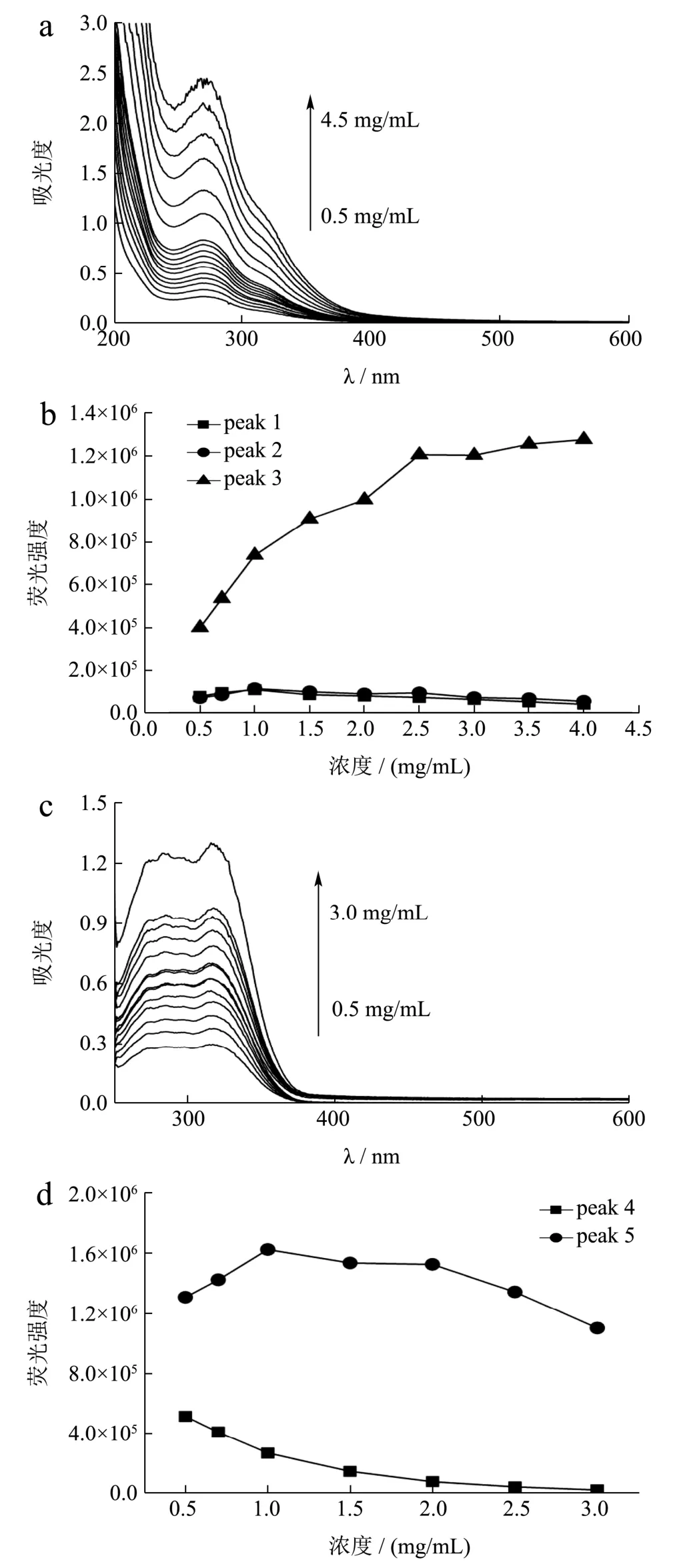
图5 不同浓度水提取物和乙酸乙酯提取物的紫外吸收光谱和荧光光谱变化 Fig.5 Absorption and fluorescence spectral changes of Angelica sinensis extracts under different concentration conditions
乙酸乙酯提取物的紫外吸收强度随供试品溶液浓度升高而增大;三维荧光光谱特征峰Peak 4 的荧光强度随浓度升高而降低,特征峰Peak 5 的荧光强度随浓度升高先增强再降低,激发波长轻微红移而发射波长位置保持不变,供试品溶液最佳检测浓度为1.00 mg/mL。
2.4 供试品溶液酸碱度的影响
重点考察了当归水提取物供试品溶液(1.00 mg/mL)的pH 值变化对三维荧光光谱的影响,结果如图6 所示。在pH 2.0~7.0 范围内,Peak l、Peak 2 和Peak 3 特征峰的峰位置基本没有变化,荧光强度随pH值增加轻微增强。在pH 7.0~12.0 范围内随pH 的增加,Peak 1荧光强度明显增加且发射波长轻微红移,Peak 2和Peak 3 的峰位置基本不变但荧光强度明显减小,尤其在pH 11.20 时的荧光光谱发生显著变化。不同pH条件下的三维荧光光谱变化表明,水提取物中的荧光组分,特别是形成475 nm 发射峰的荧光组分的分子结构中可能存在易于质子化和去质子化的基团,强酸性和强碱性条件对其荧光光谱有较大影响。当归水提取物供试品溶液在中性条件下呈现良好的三维荧光光谱特征。
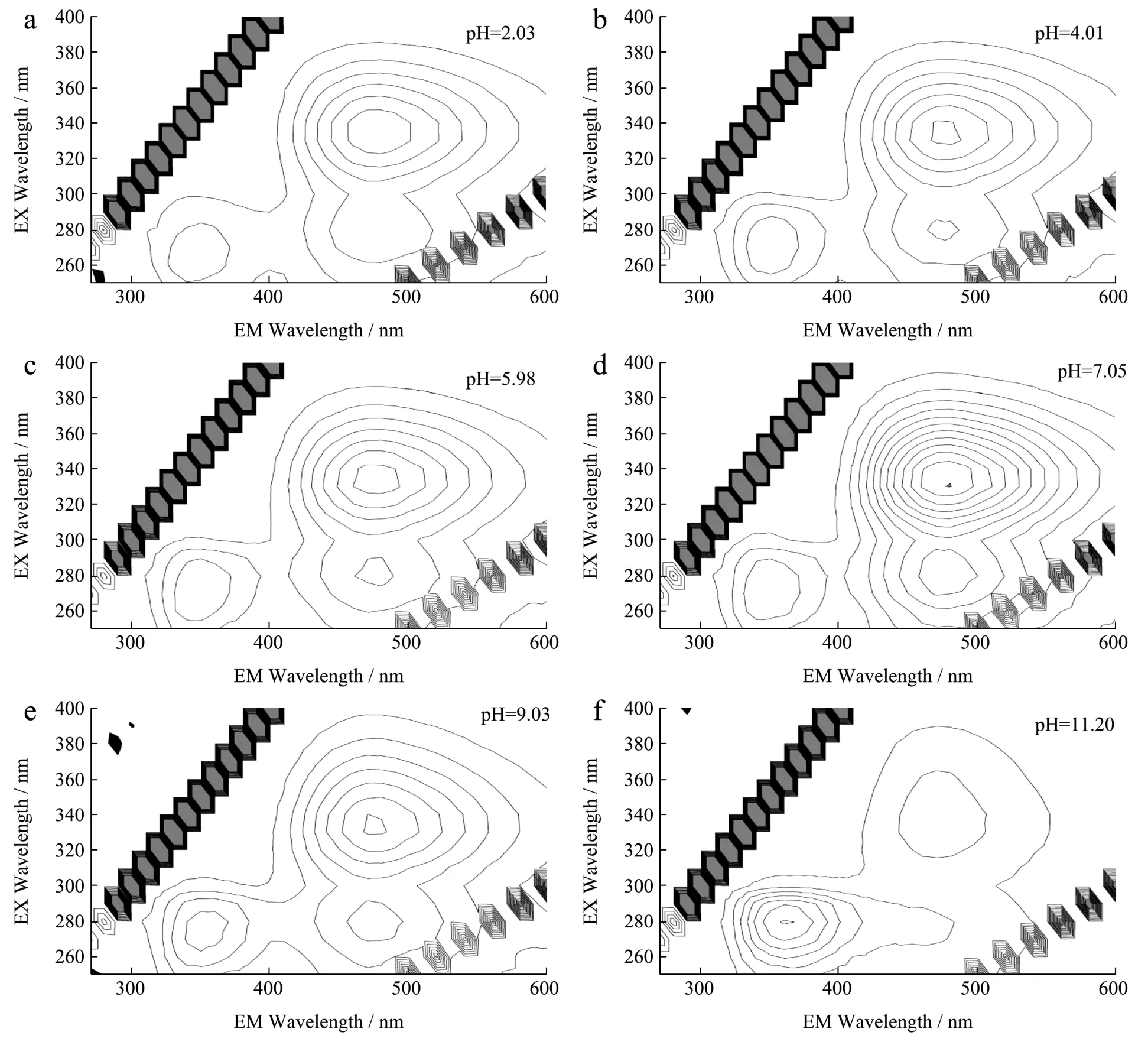
图6 不同pH 条件下的当归水提取物三维荧光光谱 Fig.6 Three-dimensional fluorescence spectra of Angelica sinensiswater extract (1.00 mg/mL) under different pH conditions
2.5 当归三维荧光光谱的专属性
采用“1.3.1.1”的供试品制备方法,制备当归同科属植物川穹、独活、北沙参、蛇床子、小茴香的水提取物,并在相同的供试品浓度和荧光光谱分析条件下分别测定三维荧光光谱,结果如图7 所示。当归与川芎的水提取物三维荧光光谱形貌非常相似,但与其他同属科中药材水提取物的三维荧光光谱在特征荧光峰的个数、位置及荧光强度等方面都存在明显差异,当归与独活、北沙参、蛇床子、茴香可以通过水提取物的三维荧光图谱进行快速鉴别,但不能有效区分当归与川穹。这一结果表明当归与川穹的水溶性提取物的物质组成和成分含量具有高度相似性。

图7 当归与同科属中药材的水提取物三维荧光光谱(1.0 mg/mL) Fig.7 Three-dimensional fluorescence spectra of the water extracts from Angelica sinensisand other umbelliferae plant (1.0 mg/mL)
采用“1.3.1.3”的供试品制备方法,制备当归同科属植物川穹、独活、北沙参、蛇床子、小茴香的乙酸乙酯提取物。在相同的供试品浓度和荧光光谱分析条件下分别测定三维荧光光谱,结果如图8 所示。当归与同科属植物的乙酸乙酯提取物的三维荧光光谱呈现不同的图谱形貌,特别是当归与川芎的特征荧光峰形貌及其位置和荧光强度具有显著差异,该结果表明当归与川穹的脂溶性成分存在较大差异,能够通过乙酸乙酯提取物的三维荧光图谱进行快速鉴别。

图8 当归与同科属中药材乙酸乙酯提取物的三维荧光光谱(1.0 mg/mL) Fig.8 Three-dimensional fluorescence spectra of the ethyl acetate extracts from Angelica sinensis and other umbelliferae plant (1.0 mg/ mL)
2.6 不同产地当归三维荧光光谱判别分析
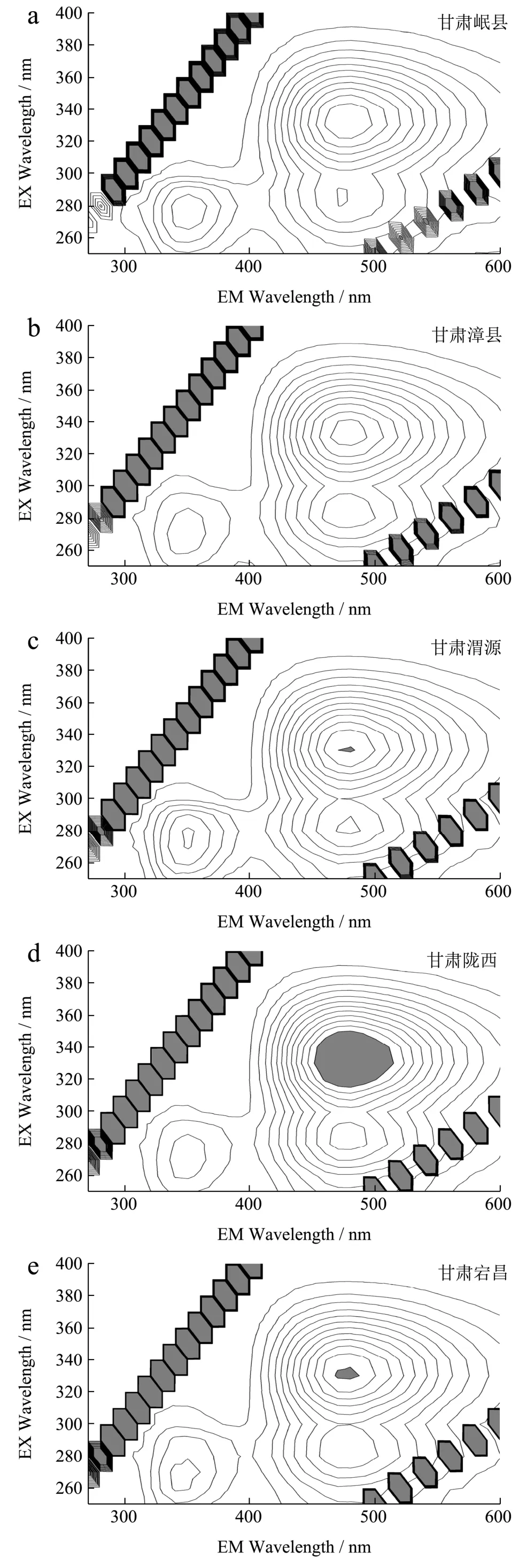
采用“1.3.1.1”制备方法,制备不同产地当归样本的水提取物供试品溶液(平行制备3 份),并在相同的光谱分析条件下,分别测定三维荧光光谱,见图9。

图9 不同产地当归的三维荧光光谱图 Fig.9 Three-dimensional fluorescence spectra of water extract ofdifferent Angelica sinensis samples from different producing areas
当归水提物三维荧光光谱图是提取物中所有荧光组分的光谱叠加,其图谱特征是各荧光物质的总体反映,直观表现为荧光光谱的最大激发波长(λex)、最大发射波长(λem)以及对应的最大发射强度(Iem)同时变化的信息。不同当归样品水提取物三维荧光光谱呈现的三个特征峰Peak 1、Peak 2 和Peak 3 的位置(λex/λem)基本一致,但发射强度(Iem)以及特征峰之间的相对强度存在差异。提取荧光光谱图的特征信息λex、λem和Iem,以各当归样品特征峰Peak 1、Peak 2 和Peak 3 对应的荧光强度Iem(三次平行测定的平均值)为变量,建立判别函数,采用SPSS 22 软件进行数据处理和判别分析。判别分析结果见表2。

表2 判别分析结果 Table 2 Discriminant analysis results
表中dis_1 为执行判别分析后系统对样品的分类,dis1_2、dis2_2、dis3_2、dis4_2 分别对应于各个数据被判定为第1 类、第2 类、第3 类、第4 类的概率,用于评价判别结果的准确性。判别分析结果将实验的当归样本分为四类,甘肃岷县、漳县、渭源县、陇西县、宕昌县和临潭县的当归样本判别为第1 类;甘肃天祝当归、青海湟源当归和欧当归分别被判别为第2、3 和4 类。表明采用荧光光谱特征值结合此判别方法可以快速有效的区分不同当归样品,分类结果反映了当归种植环境和品种对品质的影响。其中,甘肃岷县、漳县、渭源县、陇西县、宕昌县和临潭县的地理位置和气候条件最相近,也是中药当归的传统道地产区,质量具有一致性。此外,盲样X1 和X2 加入上述数据系统中进行判别分析,分别被归类为第3 类和第4 类,即青海湟源当归和欧当归,与实际样品情况完全一致。
3 结论
3.1 研究建立的当归特征提取物的三维荧光光谱呈现出特征光谱形貌,能够总体反映提取物中荧光物质信息。当归水提取物与50%甲醇提取物的三维荧光光谱具有相同的特征荧光峰,但特征荧光峰相对强度存在明显差异,表明了这两种提取物中荧光组分含量存在差异。脂溶性提取物具有完全不同于水提取物和50%甲醇提取物的三维荧光光谱特征,表明其不同的荧光物质组成。当归水提物和乙酸乙酯提取物的三维荧光光谱具有良好的专属性,能够实现当归与其同科属常用中药材的快速有效鉴别。与紫外光谱、红外光谱、高效液相色谱等分析方法相比较,在最佳提取条件下,当归提取物三维荧光光谱反映了当归药材中荧光物质成分的整体信息,所需样品量少,方便快捷。
3.2 利用建立的当归水提取物三维荧光光谱并结合计算机模式识别方法,对不同当归样本进行检测和判别分析,实现了对不同产区和品种当归的判别分类和质量一致性评价,为深入研究和建立当归三维荧光光谱指纹图谱质控技术提供参考。
猜你喜欢
煤炭与化工(2024年2期)2024-03-30 08:09:52
中老年保健(2022年2期)2022-08-24 03:20:44
中老年保健(2021年5期)2021-12-02 15:48:21
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
中成药(2018年12期)2018-12-29 12:26:02
现代营销(创富信息版)(2018年2期)2018-08-15 00:45:27
中成药(2018年6期)2018-07-11 03:01:04
中成药(2017年8期)2017-11-22 03:19:25
中成药(2017年10期)2017-11-16 00:50:27
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
